Co与甜菜碱类衍生物形成的两种配合物的结构和磁性特征
2016-05-03王学玲李松林刘雪佳
王学玲李松林*,刘雪佳
(1天津大学理学院化学系,天津 300354) (2北京航空材料研究院,北京 100095)
王学玲1李松林*,1刘雪佳2
(1天津大学理学院化学系,天津300354) (2北京航空材料研究院,北京100095)
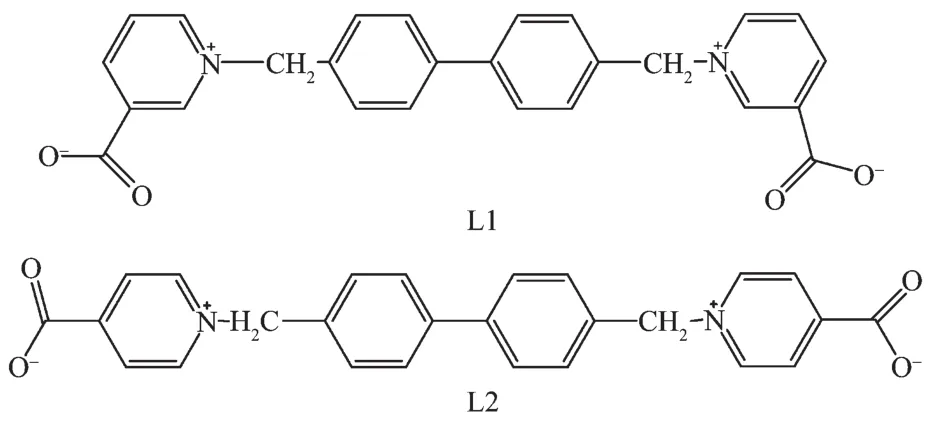
摘要:合成了2种新的Co的配合物{[Co(L1)2(H2O)2](ClO4)2·4H2O}n(1)和{[Co(L2)2(H2O)4](ClO4)2·8H2O·L2}n(2)(L1=1,1′-(4,4′-二亚甲基联苯)二(1-吡啶鎓-3-羧酸盐),L2=1,1′-(4,4′-二亚甲基联苯)二(1-吡啶鎓-4-羧酸盐)),并且分别用红外光谱、热差分析、X射线粉末衍射和X射线单晶衍射表征了这两种化合物。晶体结构分析表明L1,L2,1和2分别属于P21/c,P21/c,P1和P1空间群。化合物1中的Co与L1配位形成二维的配位聚合物,2中的Co与L2配位形成独立的阳离子。二者的晶体结构都含有大量的氢键作用或者配位键形成的孔状结构。此外,磁性研究表明,1和2中都存在反铁磁性相互作用。
关键词:甜菜碱衍生物;Co配合物;晶体结构;化学磁性
0 Introduction
In the past decades, coordination polymers have been the central research field of coordination chemistry and attracted much attention of researchers because of their fascinating structures, potential applications in magnetism, luminescence, adsorption, and catalysis, and so on[1-6]. Among these applicationsthe magnetic properties of coordination polymers occupy a special position, and many new single molecule magnets (SMMs) based on Mn12[7], Ni12[8], and to a lesser extent, Cohave been reported[9-10].
Scheme 1
1 Experimental
1.1 Materials and physical measurements
All chemicals and solvents were reagent grade without further purification. The 4,4′-bis(chloromethyl) biphenyl was synthesized according to method in the literature[17].1H NMR spectra were recorded with Bruker AVANCEⅢspectrometer operated at 400 MHz (1H). The IR spectra were recorded from KBr pellets in the range of 4 000~400 cm-1with a Bio-Rad Excalibur FTS 3000 spectrometer. Elemental analyses were performed on a Vario Micro cube element analyzer. Powder X-ray diffraction (PXRD) patterns were taken on a Rigaku D/Max-2500 X-ray diffractometer with Cu Kα radiation (λ=0.154 18 nm), and the PXRD patterns were examined out by the simulations of the single-crystal diffraction data by the Mercury program. The magnetic measurements were carried out with a Quantum Design MPMS-XL7 and a PPMS-9 ACMS magnetometer. The diamagnetic corrections for the coordination compounds were estimated using Pascal′s constants, and magnetic data were corrected for diamagnetic contributions of the sample holder. The thermal properties of 1 and 2 were investigated by thermogravimetric analysis (Fig.S1) under a nitrogen protective atmosphere, and the thermal degradation process was studied from 40 to 800℃at a heating rate of 10.0℃·min-1.
1.2 Synthesis
1.2.1 Synthesis of 1,1′-(4,4′-bis(methylene)biphenyl) bis(1-pyridinium-3-carboxylate) (L1)
The 4,4′-bis(chloromethyl)biphenyl (3.0 g, 12.0 mmol) was dissolved in ethanol (75 cm3), and ethyl nicotinate (5 cm3, 36.7 mmol, excessive 50%) was added. The reaction mixture was refluxed for 48 h and the solvent was removed under reduced pressure. Then the syrupy product was dissolved in acetone, and white precipitate formed under continuously stirring of the solution. The precipitate was filteredand washed with acetone until the filtrate being colorless. The product was dissolved in HCl (15%, 60 cm3) and refluxed for 4 h. After removal of the solvent under reduced pressure, the residue was dissolved in minimum amount of water and treated with 1,2-epoxypropane at 30℃until pH≈7. The solvent was removed under reduced pressure, then the syrupy residual was dissolved in DMF, and the mixture was stirred until precipitate formed. After recrystallization of the precipitate in H2O/DMF, the product was obtained. Yield: 2.8 g, 41.8%. IR (cm-1): 3 458 (m), 3 067(m), 1 647(s, carboxylate νasym), 1 374 (s, carboxylate νsym), 778(s), 619(m);1H NMR (400 MHz, D2O):δ 9.04 (s, 2H), 8.77 (d, J=6.1 Hz, 2H), 8.61 (d, J=8.0 Hz, 2H), 7.91~7.78 (m, 3H), 7.24 (d, J=8.2 Hz, 4H), 7.13 (d, J=8.1 Hz, 4H), 5.67 (s, 4H), 2.91 (s, 3H), 2.76 (s, 3H). Elemental analysis Calcd. for C26H20N2O4· 3H2O·DMF(%): C, 63.16; H, 5.99; N, 7.62. Found(%): C, 63.10; H, 5.89; N, 7.58.
The synthesis procedure of L2 was similar to that of L1 except that ethyl isonicotinate was used instead of ethyl nicotinate. Yield: 3.2 g, 52.3%. IR (cm-1): 3 423(s), 3 041(m), 1 634(s, carboxylate νasym), 1 368(s, carboxylate νsym), 774(m), 694(m);1H NMR (400 MHz, D2O)δ 8.89 (d, J=6.7 Hz, 4H), 8.16 (d, J=6.7 Hz, 4H), 7.64 (d, J=8.2 Hz, 4H), 7.46 (d, J=8.2 Hz, 4H), 5.78 (s, 4H). Elemental analysis Calcd. for C26H20N2O4·4H2O (%): C, 62.90; H, 5.65; N, 5.65. Found(%): C, 63.20; H, 5.73; N, 5.58.
1.2.2 {[Co(L1)2(H2O)2](ClO4)2·4H2O}n(1)
Co(ClO4)2·6H2O (366.0 mg, 1.0 mmol) and L1· 3H2O·DMF (275.5 mg, 0.5 mmol) were dissolved in 8 cm3H2O/DMF (2∶1, V/V), and the result solution was heated at ca. 80℃under stirring for 30 min. The reaction mixture was cooled to room temperature and filtered. Pink prismatic single crystals suitable for single crystal X-ray diffraction structure analysis were obtained after the filtrate was allowed to stand for several days at ambient temperature. Yield: 512 mg, 42.2%. IR(cm-1): 3 396(s), 3 081(m), 1 640(s, carboxylate νasym), 1 388 (s, carboxylate νsym), 1 089 (s), 764 (m), 684 (m), 625 (m). Elemental analysis Calcd. for C52H52Cl2CoN4O22(%): C, 51.37; H, 4.30; N, 4.61. Found (%): C, 50.77; H, 4.67; N, 4.40.
1.2.3 {[Co(L2)2(H2O)4](ClO4)2·8H2O·L2}n(2)
The preparation procedure of this compound was analogous to that of 1. To the aqueous solution of Co(ClO4)2·6H2O (366 mg, 1.0 mmol) and L2·4H2O (248 mg, 0.5 mmol), 8 cm3H2O/DMSO (2∶1, V/V) was added dropwise. Pink prismatic crystals were obtained. Yield: 700 mg, 40.1%. IR(cm-1): 3 410(s), 3 051(m), 1 632(s, carboxylate νasym), 1 380 (s, carboxylate νsym), 1 088(s), 856(m), 624(m). Elemental analysis Calcd. for C78H84Cl2CoN6O32(%): C, 53.57; H, 4.81; N, 4.81. Found(%): C, 53.22; H, 5.17; N, 4.71.
1.3 X-ray diffraction data collection, structure determination and refinement
The intensities of L1·3H2O·DMF, L2·4H2O, 1 and 2 were collected on a Rigaku Saturn CCD diffractometer (confocal-monochromated Mo Kα radiation: λ=0.071 073 nm). Absorption correction based on multi-scan was applied to the intensity data processing.
The structures were solved by direct method using SIR2014 software[18], and the non-hydrogen atoms except the oxygen atoms of the disordered perchlorate anion in 2 were refined anisotropically by full-matrix least squares method based on all the diffraction data using the SHELX 2014 program package[19]. Attempting to anisotropically refine the oxygen atoms of the disordered perchlorate anion in 2 always causes some oxygen atoms non-positively determined, thus isotropically refinements are applied to them. All hydrogen atoms unless those on coordination free water molecules were placed at their calculated positions, however, the hydrogen atoms on water molecules were located by calculation using Wingx[20]or from the difference Fourier map, and allowed to ride on their respective parent atoms and included in the structure factor calculations. One of the benzene rings in 1 adopts two orientations with the occupation of 0.471 and 0.529, respectively, and a perchlorate anion in 2 displays two-fold disorder about Cl2-O20 bond with the occupation of 0.456 and 0.544, respectively. Checking the structure of 2 with Platon program shows it may crystallize in space group P1, however, transforming the structure from space group P1 to P1with Platon and refining it using SHELX 2014 lead to unacceptable R factors and abnormal thermal parameters. The existence of disordered perchlorate ion in 2 may be responsible for the structure only solved and refined in space group P1.
CCDC: 1439325, L1·3H2O·DMF; 1442802, L2· 4H2O; 1439327, 1; 1439328, 2.
2 Results and discussion
2.1 IR spectra of the compounds
The IR spectra of L1·3H2O·DMF, L2·4H2O, 1 and 2 were measured. The absorptions at 1 647, 1 634, 1 640, 1 632 cm-1are assigned to the asymmetric stretching vibration νasym, and the absorptions at 1 374, 1 368, 1 388, 1 380 cm-1correspond to the symmetric stretching vibration νsymof the carboxylate groups in L1·3H2O·DMF, L2·4H2O, 1 and 2, respectively. It is well known that the Δν value (Δν=νasym-νsym) of carboxylate group is relative to its coordination mode with metal ions[21]. The similar Δν values (273, 266, 252, 252 cm-1for L1·3H2O·DMF, L2·4H2O, 1 and 2, respectively) indicate that the environments of the carboxylate groups of these compounds in solid state have little difference, which implies that the hydrogen bond formation have a significant effect on the vibration of the carboxylate group.
2.2 Crystal structure analysis
2.2.1 Crystal structure of L1·3H2O·DMF
Compound L1·3H2O·DMF crystallizes in space group P21/c with the asymmetric unit consisting of one L1 molecule, one DMF molecule and three water molecules. The L1 molecule exhibits a Z-conformation(relative to biphenyl group) with the pyridinium rings being nearly perpendicular to the phenyl rings (106.77(7)°and 106.02(6)°) (Fig.1). Adjacent L1 molecules and two water molecules are joined by hydrogen bonds (O…O distances: 0.281 7(2)~0.288 1(2) nm) to zig-zag chains with nodes of hydrogen bonded 8-membered rings in chair conformation. Sinusoidal planes are constructed by gluing adjacent chains through hydrogen bonds between oxygen atoms of carboxylate groups and O3W′s molecules (O…O distances: 0.272 5(2) and 0.269 3(2) nm) (Fig.2), and DMF molecules are fitted in between sinusoidal planes. Although there are four phenyl and pyridinium rings in the compound, no π-π interactions are found in L1·3H2O·DMF in solid state.

Table1 Crystallographic and data collection parameters for L1·3H2O·DMF, L2·4H2O, 1 and 2
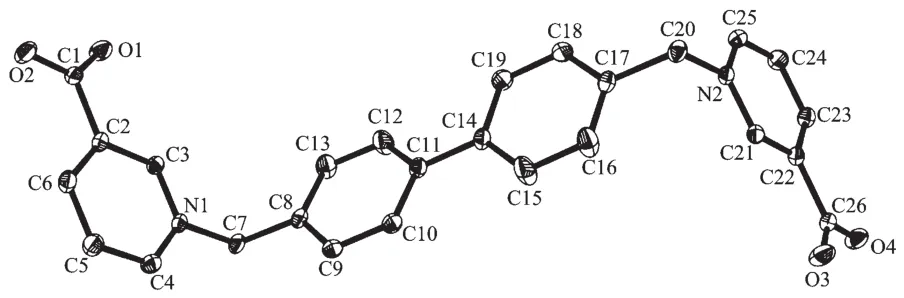
Hydrogen atoms are omitted for clarity; Thermal ellipsoids are drawn at 45% probability levelFig.1 Molecular structure of L1

Symmetry code: A: x-1, y, z-1Fig.2 Sinusoidal plane in L1·3H2O·DMF
2.2.2 Crystal structure of L2·4H2O
L2 together with four water molecules crystallizes in the space group P21/c. Similar to L1, the L2 molecule also adopts a Z-conformation relative to the biphenyl group. However, the dihedral angles between the pyridinium ring and the adjacent phenyl ring being 88.51(8)°and 90.40(8)°indicate that they are more close to mutually perpendicular (Fig.3). Like that in 1, adjacent L2 molecules and water molecules are linked by hydrogen bonds (O…O distances: 0.273 2(3)~ 0.279 5(3) nm) to zig-zag chains with 8-membered hydrogen bonded rings in chair conformation running down a axis. The hydrogen bonded rings can be classified into two types. One of which forms 3-membered hydrogen bonded rings with O4W′s (O…O …O angles: 55.58(6)°~63.73(6)°, O…O distances: 0.279 5(3)~0.303 9(6) nm), and the other links to adjacent chains through hydrogen bonding to a pair of water molecules (O3W′s) (O…O…O angle: 107.55(5)°, O…O distances: 0.279 2(4) and 0.308 2(4) nm), where O2W′s act both as bi-hydrogen bonding donors and bi-hydrogen bonding acceptors, thus forming hydrogen bonded planes (Fig.4). It is worthy to note that there exist π-π interactions between pyridinium and phenyl rings (centroid-centroid distance: 0.372 6(2) nm, shift distance: 0.132 7(3) nm) of the screw-axis related adjacent planes, which further stabilize the 3D structure of the compound.
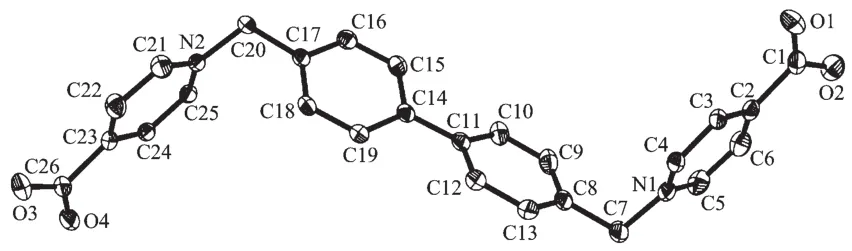
Hydrogen atoms are omitted for clarity; Thermal ellipsoids are drawn at 45% probability levelFig.3 Molecular structure of L2
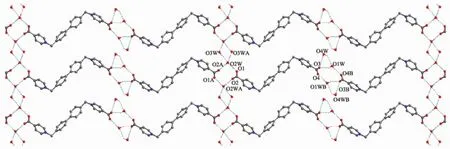
Symmetry code: A: -2-x, 4-y, 1-z; B: 1-x, 4-y, 2-zFig.4 Hydrogen bonded planes in L2·4H2O
2.2.3 Crystal structure of 1
As depicted in Fig.5, the Coatom locates at the crystallographic inverse center, and is six coordinated in a MO6octahedral sphere, in which the four oxygen atoms (O1, O1A, O3, O3A) from monodentate carboxylate groups (Co-O distances: 0.209 1(2) and 0.209 5(2) nm) lay on the equatorial plane, and the two oxygen atoms (O1W, O1WA) from aqua ligands occupy axial positions (Co-O distance: 0.212 5(2) nm). Each of the aqua ligands forms hydrogen bonds with a pair of uncoordinated oxygen atoms of the carboxylate groups. Although the coordination octahedron is elongated somewhat along axial directions, it isobviously in a nearly perfect octahedral conformation (O…Co…O angles: 86.89(9)°~93.11(9)°). Each Coatom is connected to nearby four Coatoms through four L1 molecules thus leading to the formation of coordination plane with large holes (Fig.6). The hole is formed by 84-membered ring with a dimension of 1.892 5(6) nm (Co…Co)×1.587 8(4) nm (Co…Co). Three planes which are shifted along a axis by a relative to each other pack together to form a layer. By careful analysis, there are no obvious interactions, either hydrogen bonding or π-π interactions, found between these planes. The distance between adjacent layers is 1.341 1 nm, and the minimum distance between Coatoms is 0.824 8(3) nm.

Table2 Selected bond lengths (nm) and angles (°) for L1·3H2O·DMF and L2·4H2O

Hydrogen atoms are omitted for clarity; Thermal ellipsoids are drawn at 45% probability level; Symmetry code: A: -x+1, -y+1, -z+1Fig.5 Coordination environment of Coin 1
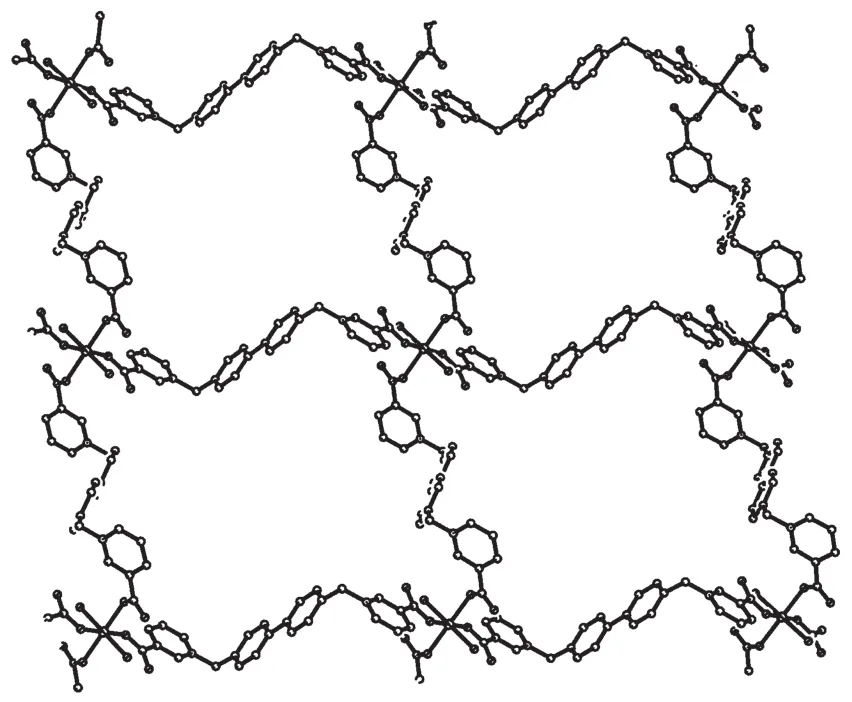
Fig.6 Plane with holes in 1
2.2.4 Crystal structure of 2
Analogous to that in 1 the Coatoms are also in an octahedral MO6coordination environment (Fig. 7), however, the Coatoms do not locate at crystallography inverse centers. Furthermore the equatorial coordination positions are occupied by a pair of transrelated monodentate carboxylate groups and a pair of trans-related aqua ligands, which forces the MO6coordination sphere in 2 being more distorted from perfect octahedron than that in 1 (Co-O distances: 0.205 4(3)~0.215 6(3) nm, O-Co-O angles: 83.6(1)°~ 100.7(1)°). Unlike that in 1, the cation which is discrete rather than coordination polymer contains a pair of L2 molecules with only two of their four carboxylate groups being involved in coordination to the metal ion, and two coordination free carboxylate groups positioning at the ends of the cation as shown in Fig. 7. As hydrogen bond accepters, one of the oxygen atoms from each coordination free carboxylate group link to the aqua ligands (O…O distances: 0.267 4(4) and 0.277 8(4) nm) of the adjacent cations in the direction (111), and a chain, which is along the (111)direction, with quasi-parallel quadrilateral rings (48-membered, 1.672 9 (0) nm (Co1…Co1A)×1.538 4(4) nm (C7…C46A)) is formed (Fig.8). The other oxygen atoms of the coordination free carboxylate groups on the chain attach to the aqua ligands of the adjacent chains thus extending the structure to a layer of channels running down the a direction (Fig.9).
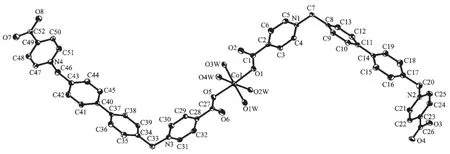
Hydrogen atoms are omitted for clarity; Thermal ellipsoids are drawn at 45% probability levelFig.7 Coordination environment of Coin 2
In spite of the coordinating L2 molecules, the crystal of 2 also contains coordination free L2 molecules, which are connected by hydrogen bonds (O …O distances: 0.273 8(5)~0.279 1(5) nm) between the carboxylate groups and water molecules to form a zig-zag thread (Fig.10). The conformation of L2 is nearly the same with that in L2·4H2O, which implies that it may be the most stable conformation of L2 molecule. It is interesting to note that the thread penetrates the channels of different layers, and interacts with channels through hydrogen bonds (O…O distances: 0.257 3(5)~0.287 9(5) nm) and π-π interactions (centroid-centroid distance: 0.389 7(3) and 0.385 8(3) nm, shift: 0.146 1(8) and 0.131 0(7) nm, respectively), weaving the channeled layers to a hydrogen bonded and π-π stacked 3D structure (Fig.11).
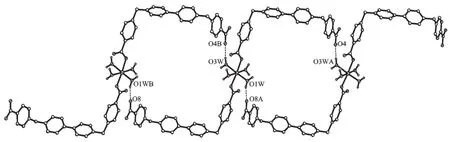
Symmetry code: A: x+1, y+1, z+1; B: x-1, y-1, z-1Fig.8 48-membered quasi-parallel quadrilateral rings in a chain

Symmetry code: A: x+1, y+1, z+1; B: x+2, y+1, z+1; C: x-1, y, zFig.9 Layer of channels in 2
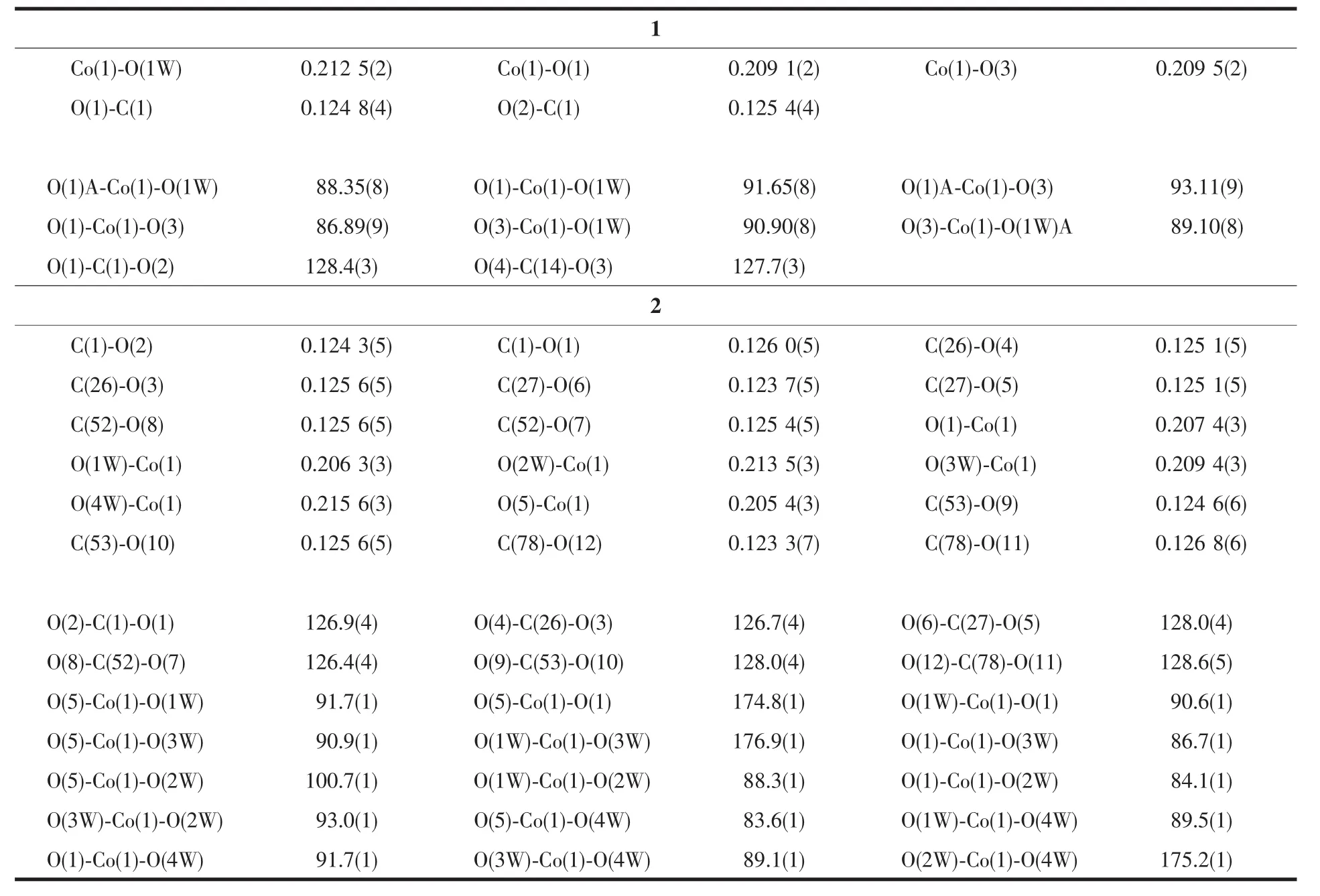
Table3 Selected bond lengths (nm) and angles (°) for 1 and 2
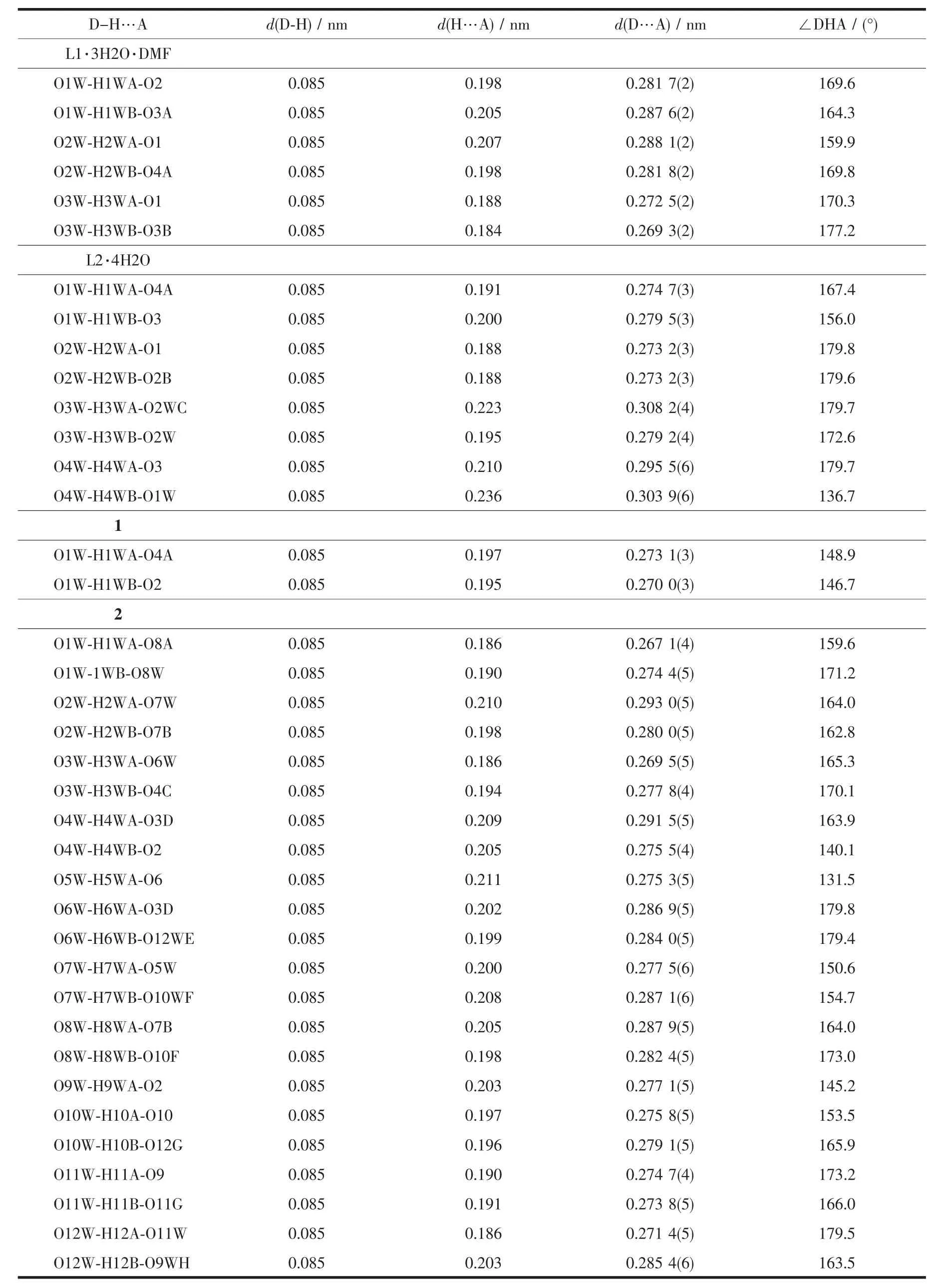
Table4 Selected bond lengths and bond angles of hydrogen bonds in L1·3H2O·DMF, L2·4H2O, 1 and 2

Symmetry code: A: x+2, y, z+1Fig.10 zig-zag thread constructed by coordination free L2 molecules in 2

Fig.11 Threads penetrate the channels of different layers in 2
2.3 Powder X-ray diffraction results
The powder X-ray diffraction (PXRD) experiments were carried out on the as-synthesized samples. As can be seen in Fig.S2, the experimental PXRD patterns and the simulated patterns generated from the single-crystal diffraction data of L1·3H2O·DMF, L2· 4H2O, 1 and 2 are in good agreement, which indicates that each compound obtained is in single phase state.
2.4 Thermogravimetric Analyses of 1 and 2
The TG curve of 1 exhibits four main steps of weight losses occurring in the range of 30~130℃, 130~193℃, 233~280℃and 280~680℃, which corresponds to the weight losses of free water molecules (Obsd. 5.82%, Calcd. 5.93%), aqua ligands (Obsd. 2.85%, Calcd. 2.96%), the ClO4-anions (Obsd. 16.30%, Calcd. 16.38%) and the completely decomposition of the organic ligands, respectively. Although the residuals are known as cobalt oxides, their composition can′t be identified. The TG curve of 2 shows three main steps of weight losses. Firstly, it losses twelve water molecules from 30 to 105℃(Obsd. 12.30% , Calcd. 12.36% ). The dehydrated compound remains stable up to 230℃and then upon further heating the weight loss of 39.30% (Calcd. 39.60%) from 230 to 310℃tallies with the loss of two ClO4-anions and one coordination free ligand. Then upon further heating, it decomposes to unidentified products which are speculated as some oxides of the cobalt.
2.5 Magnetic studies of 1 and 2
The direct-current (dc) magnetic susceptibility of 1 and 2 have been carried out in an applied magnetic field of 1 kOe in the range of 2~300 K. The plots of χMT versus T, where χMT is the molar magnetic susceptibility, are shown in Fig.12. Upon cooling, the χMT values of 1 remain almost unchanged between 300 and 150 K. When temperature further decreases, the χMT product decreases slowly to reach a minimum value of 1.96 cm3·mol-1·K at 2 K. For 2, the χMT values keep nearly unvaried between 300 and 100 K and reach a minimum value of 1.82 cm3·mol-1·K at 2 K. The falling trends can be ascribed to the antiferromagnetic interactions among the Coatoms in compounds 1 and 2[22]. At room temperature the χMT values of 1 and 2 are 2.91 cm3·mol-1·K (μeff=4.82μB) and 2.96 cm3·mol-1·K (μeff=4.86μB), respectively, both of which are higher than that expected for one highspin d7ion (1.87 cm3·mol-1·K,μeff=3.87μB, g=2.0), but are close to the value expected for independent spin and orbital moments (L=3, S=3/2,μLS=[L(L+1)+4S(S+ 1)]1/2=5.2μB). It is well known that when coordinated the orbital and spin coupling of Coatom at high temperature would produce effect magnetic moments in the range of 4.7μB~5.2μBfor CoX6coordination sphere, and the effects on magnetic moments vary with how much of the distortion of CoX6sphere from octahedron[23-25]. In 1 and 2, the Coatoms are both in CoO6coordination spheres, but they are distorted from octahedron in different extent, and this may be responsible for the different values of χMT at roomtemperature. In addition, the differences in the minimum Co…Co distances (0.824 8(3) nm for 1 and 0.907 5(2) nm for 2) and the pore sizes (1.892 5(6) nm (Co…Co)×1.587 8 (4) nm (Co…Co) for 1 and 1.672 9(0) nm (Co1…Co1A)×1.538 4(4) nm (C7…C46A) for 2) may also have some influence on the distinctions of their magnetism[13,26-27]. The relations of χMT vs T data for both 1 and 2 show Curie-Weiss behaviors in the range of 100~300 K, with C=2.98 cm3·K·mol-1and θ=-7.99 K for 1, and C=3.04 cm3· K·mol-1and θ=-7.27 K for 2, respectively. The negative values of θ for both compounds also indicate an antiferromagnetic interaction in 1 and 2.
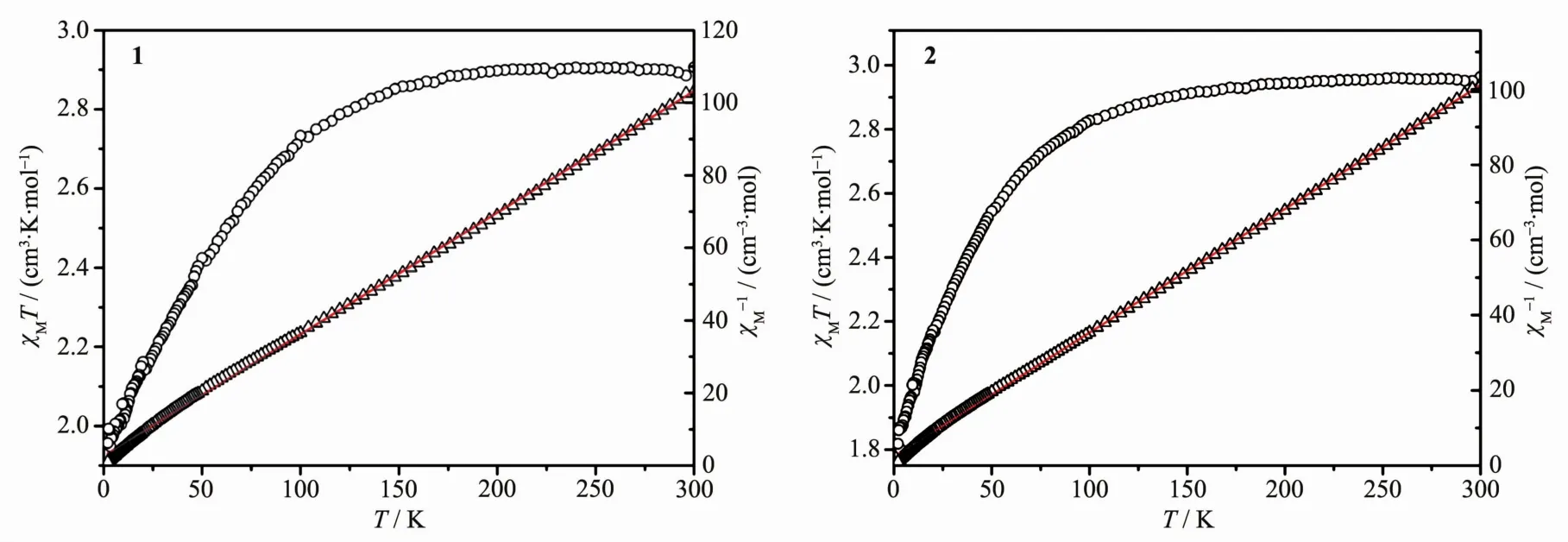
Fig.12 Plots of χMT vs T (circles) and χM-1vs T (triangles) for 1 and 2
3 Conclusions
Two new bidentate betaine derivatives 1,1′-(4,4′-bis(methylene)biphenyl)bis(1-pyridinium-3-carboxylate) (L1) and 1, 1′-(4, 4′-bis(methylene)biphenyl)bis(1-pyridinium-4-carboxylate) (L2) have been synthesized. Both the molecules of L1 and L2 contain four aromatic rings and two naked carboxylate groups which occupy the ends of the molecule and are separated by an 18-membered atomic chain. The connective -CH2groups in the molecules enable it readily adjusting its conformation to accommodate its coordination environment. All these characteristics of the compounds enable them to suitably construct coordinated or hydrogen bonded networks with large pores. In addition, the plenty of aromatic rings in the molecules can be involved in π-π interactions, which could contribute certain stability to the compounds in solid state.
Supporting information is available at http://www.wjhxxb.cn
References:
[1] Yin F, Chen J, Liang Y F, et al. J. Solid State Chem., 2015, 225:310-314
[2] Zhao M X, Xiong L Q, Qi C M. J. Inorg. Organomet. Polym., 2015,25:1239-1249
[3] Sun Y Q, Liu S Y, Xu Y Y, et al. Polyhedron, 2014,74:39-48
[4] Raptopoulou C P, Psycharis V. Inorg. Chem. Commun., 2008, 11:1194-1197
[5] Maguerb P. L, Ouahab L, Golhen J S, et al. Inorg. Chem., 1994,33:5180-5187
[6] GUO Hai-Fu(郭海福), LEI Jia-Mei(雷佳眉), MA De-Yun(马德运). Chinese J. Inorg. Chem.(无机化学学报), 2015,31 (12):2385-2392
[7] Sessoli R, Gatteschi D, Caneschi A, et al. Nature, 1993,365: 141-143
[8] Cadiou C, Murrie M, Paulsen C, et al. Chem. Commun., 2001,37:2666-2667
[9] Wang L F, Li C J, Chen Y C, et al. Chem. Eur. J., 2015,21: 2560-2567
[10]Greg B, Ekk S. Inorg. Chem., 1985,24:4580-4584
[11]Carlin R L, Dekker C. Transition Metal Chemistry. New York: Marcel Dekker, Inc., 1965.
[12]Desiraju G R. Crystal Design: Structure and Function. Chichester: John Wiley & Sons, Ltd. 2003.
[13]Murray K S. Eur. J. Inorg. Chem., 2008:3101-3121
[14]Kostakis G E, Perlepes S P, Blatov V A, et al. Coord. Chem. Rev., 2012,256:1246-1278
[15]Kahn O. Molecular Magnetism. New York: VCH Publishers, 1993.
[16]Murrie M. Chem. Soc. Rev., 2010,39:1986-1995
[17]FANG Qi(方奇), ZHU Chun-Yan(朱春燕), REN Xiao-Li(任晓丽), et al. Fine and Specialty Chemicals(精细与专用化学品), 2005,13(10):25-27
[18]Burla M C, Caliandro R, Camalli M, et al. J. Appl. Cryst., 2012,45:357-361
[19]SHELXTL Version 6. 14 8/06/00, Bruker AXS, 2000-2003.
[20]Farrugia L J. J. Appl. Crystallogr., 1999,32:837-838
[21]Nakamoto K. Infrared and Raman Spectra of Inorganic and Coordination Compounds. 6th Ed. New Jersey: John Wiley & Sons, Inc., 2009.
[22]Nemec I, Machata M, Herchel R, et al. Dalton Trans., 2012, 41:14603-14610
[23]Carlin R L. Magnetochemistry. New York: Springer-Verlag, 1986.
[24]Laget V, Hornick C, Rabu P, et al. Coord. Chem. Rev., 1998, 178-180:1533-1553
[25]Figgis B N, Hitchman M A. Ligand Field Theory and its Applications. Canada: Wiley-VCH, 2000.
[26]Leznoff D B, Lefebvre J. Gold Bull., 2005,38(2):47-54
[27]Herve K, Cardor O, Golhen S, et al. Res. Chem. Intermed., 2008,34(2/3):191-199
Crystal Structures and Magnetic Properties of Two CoCoordination Compounds Based on Bidentate Betaine Derivatives
WANG Xue-Ling1LI Song-Lin*,1LIU Xue-Jia2
(1Department of Chemistry, School of Science, Tianjin University, Tianjin 300354, China)
(2Beijing Institute of Aeronautical Materials, Beijing 100095, China)
Abstract:Two new Cocoordination compounds, namely {[Co(L1)2(H2O)2](ClO4)2·4H2O}n(1) and {[Co(L2)2(H2O)4] (ClO4)2·8H2O·L2}n(2) have been synthesized based on bidentate betaine derivatives (L1=1,1′-(4,4′-bis(methylene) biphenyl)bis(1-pyridinium-3-carboxylate), L2=1,1′-(4,4′-bis(methylene)biphenyl)bis(1-pyridinium-4-carboxylate)), and have been characterized by infrared spectrum, thermogravimetric analysis, powder X-ray diffraction (PXRD) and single crystal X-ray diffraction method. Single crystal X-ray analyses revealed that L1, L2, 1 and 2 crystallize in space group P21/c, P21/c, P1 and P1, respectively. The Coions in 1 coordinated with L1 molecules form 2D coordination polymer, the Coions in 2 coordinated with L2 molecules are discrete cations. The crystal structures of both compounds exhibit large hydrogen bonded or coordinated pores. Magnetic measurements indicated that there exist antiferromagnetic interactions in compounds 1 and 2. CCDC: 1439325, L1·3H2O·DMF; 1442802, L2·4H2O; 1439327, 1; 1439328, 2.
Keywords:bidentate betaine derivative; Cocoordination compound; crystal structure; chemomagnetism
DOI:10.11862/CJIC.2016.089
中图分类号:O614.81+2
文献标识码:A
文章编号:1001-4861(2016)04-0720-11
收稿日期:2015-12-23。收修改稿日期:2016-02-27。(*)通信联系人。E-mail:slli@tju.edu.cn
