藏猪、长白猪肠道微生物菌群的定量分析及比较研究
2016-04-21李江凌陈晓晖王秋实龚建军廖党金吕学斌
李江凌 陈晓晖 刘 锐 王秋实 龚建军 曾 凯 廖党金 高 荣 吕学斌
(1四川畜牧科学研究院,四川成都610066;2四川大学,四川成都610063)
藏猪、长白猪肠道微生物菌群的定量分析及比较研究
李江凌1陈晓晖1刘锐1王秋实1龚建军1曾凯1廖党金1高荣2吕学斌1
(1四川畜牧科学研究院,四川成都610066;2四川大学,四川成都610063)
摘要:为研究藏猪肠道微生物菌群特性及耐粗饲特性,本试验选择生长发育和营养状况正常的藏猪和长白猪各18头,随机分成两组即常规组和粗粮组,每组9头。分别于0、30、60日龄采集肠道内容物,应用荧光定量PCR的方法对肠道中四种细菌(大肠杆菌、乳酸杆菌、芽孢杆菌、梭状杆菌)开展定量分析及对比研究,结果显示:在出生时,藏猪、长白猪肠道内四种细菌的数量差异不显著;随着生长发育,断奶后藏猪肠道内大肠杆菌、乳酸杆菌数量显著多于长白猪(<0.05),梭状杆菌数量极显著多于长白猪(<0.01),芽孢杆菌属两个猪种之间无明显差异。饲喂粗饲料后,藏猪肠道内梭状杆菌、芽孢杆菌数量极显著增加(<0.01),而长白猪乳酸杆菌数量显著增加(<0.05)。这些结果为进一步研究藏猪、长白猪耐粗饲性状差异的遗传机理提供了科学依据。
关键词:藏猪;长白猪;耐粗饲性能;肠道微生物;荧光定量PCR
耐粗饲性状主要指生猪对饲料营养的要求低,可以适应低能量的饲料来源,比如青绿饲料、纤维素含量高的饲料等,耐粗饲的猪品种对纤维类物质的利用率较高,可以充分利用各种来源的饲料资源,缓解农业资源的供需矛盾。生猪耐粗饲的能力与其肠道特定的微生态区系密切相关。动物肠道相对稳定的微生态系统和肠道微生物间的相互稳定作用促进了特定动物复杂共生体的共同进化,并发挥了促进动物正常生长发育和健康的重要作用。因此,开展对藏猪、长白猪肠道微生物区系的研究,对于生猪耐粗性状的遗传机理研究尤为关键。
过去研究动物体内的肠道微生物通常应用传统离体培养方法,这种方法的主要缺点是对于严格厌氧菌和一些尚未得到鉴定的细菌不能被正常分离纯化,在离体培养状态下的微生物在形态和特征与在肠道自然状态下均存在显著差异。随着科技的发展,目前普遍应用不依赖微生物培养的分子生物技术研究动物胃肠道微生物区系,主要方法有应用16SrDNA、DGGE、定量PCR等,可研究土壤、动物肠道、食品微生物等的群落变化。
本试验应用荧光定量PCR技术研究藏猪、长白猪肠道中四种细菌(大肠杆菌、乳酸杆菌、芽孢杆菌、梭状杆菌)的多样性,分析常规饲养及粗粮饲养条件下藏猪、长白猪四种细菌数量的变化情况,为揭示藏猪的耐粗性状提供依据。
1 材料与方法
1.1试验动物
选择生长发育和营养状况正常的0月龄藏猪、长白猪各18头,试验前驱虫、去势,并免疫相关疫苗。
1.2试验设计
藏猪和长白猪随机分成两组即常规组和粗粮组,每组9头。参照地方猪、外种猪的饲养标准,根据本场猪群情况,以玉米和大豆为主要原料配制育肥猪基础日粮,基础日粮均为粉料。对照组饲喂基础日粮,试验组饲喂由90%的基础饲粮和10%的菊苣混合而成(菊苣切碎后与饲料混匀)的粗粮。
1.3样品采集及DNA提取
分别于0、30、60日龄屠宰猪只后采集结肠部位内容物,-20℃保存,取回实验室后用天根公司TIANamp Stool DNA Kit粪便基因组DNA提取总DNA,-20℃保存待用。
1.4肠道内常见菌群的定量检测
1.4.1荧光定量PCR标准曲线的建立
对肠道内常见的大肠杆菌、乳酸杆菌、芽孢杆菌、梭状杆菌等4种菌群做定量分析。设计引物荧光定量PCR反应的条件如表1~3。

表1 荧光定量PCR所用引物及退火温度
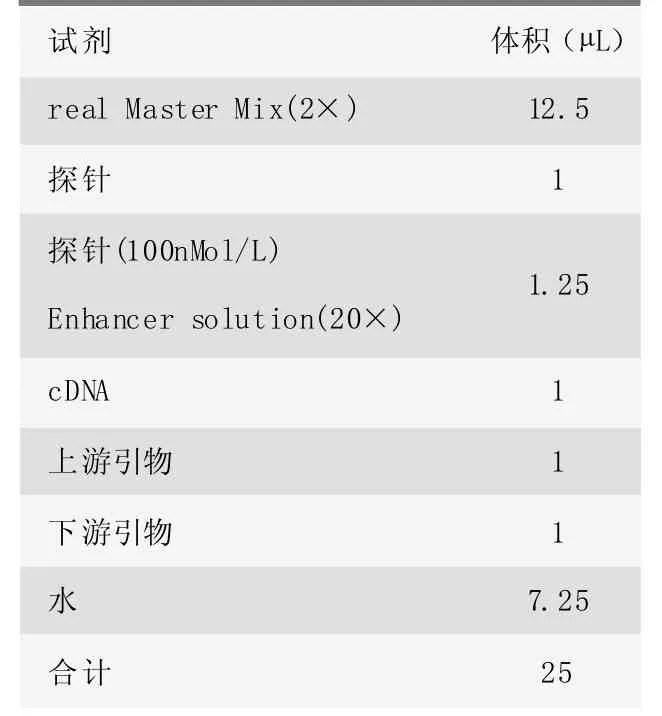
表2 实时定量PCR反应体系设置

表3 PGR扩增反应程序
采用常规方法分别完成了荧光定量标准质粒构建、建立标准曲线及线性范围的确定。
(1)使用E.Z.N.A.TM plasmid Mini Kit I提取经测序鉴定的阳性克隆重组质粒,然后用紫外可见分光光度计测定重组质粒DNA的浓度,并按如下公式换算成拷贝数。
DNA(copy)=50(μg/mL)× 10-6×OD×NA(阿佛加德罗常数:6.02×1023分子/摩尔)/660(碱基对平均分子量)×扩增碱基数
(2)标准曲线的制作是在Real-time PCR的检测范围内,以各浓度梯度起始拷贝数的对数值为X轴,相应的Ct值为Y轴作回归曲线,得出Real-time PCR标准曲线的表达式。
1.4.2样品的Real-time PCR测定
对上述重组质粒DNA构建的标准曲线样品和各DNA样品进行荧光定量PCR检测。每个样品作3次重复,采用TaqMan Real-Time PCR Premix Kit试剂盒于BiONEER公司ExicyclerTM96实时荧光定量PCR进行绝对定量PCR分析。荧光定量PCR反应体系和反应条件同标准曲线的建立。根据相应的标准曲线和相应的Ct值,计算出各个样品中4种菌群的拷贝数和每克粪便中的数量。
1.5数据整理分析
Real-time PCR结果采用SPSS 21.0进行显著性分析。
2 结果分析
2.1标准曲线及方程
四个菌属实时荧光定量PCR标准曲线,重复管Ct值之差在0.5以内,以1×109~1×104拷贝数/μL标准品阳性模板进行荧光定量,连续10倍梯度浓度之间Ct值之差在3.33左右;以初始标准品模板量的对数为横坐标,Ct值为纵坐标,分别绘制出4种菌属的荧光定量PCR标准曲线方程,结果见表4。
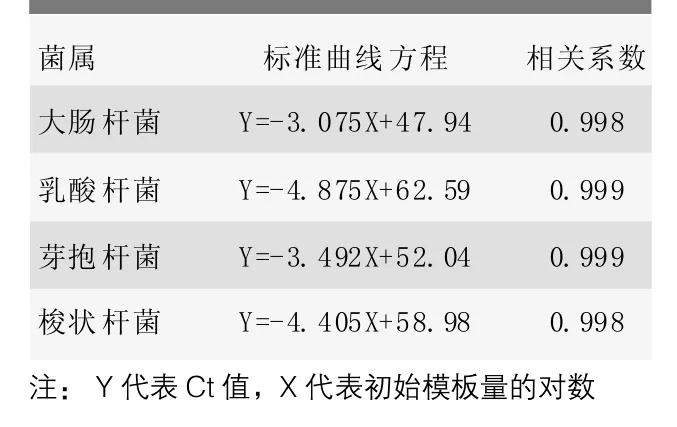
表4 4个菌属的标准曲线方程
2.2实时荧光定量检测藏猪和长白猪粪便中4种菌群
使用已经建立起来的标准曲线和方法对试验藏猪、长白猪粪便DNA中4种菌属数量进行检测。由于两样本的总体方差d2均未知,且为小样本,用t检验,结果如表5。
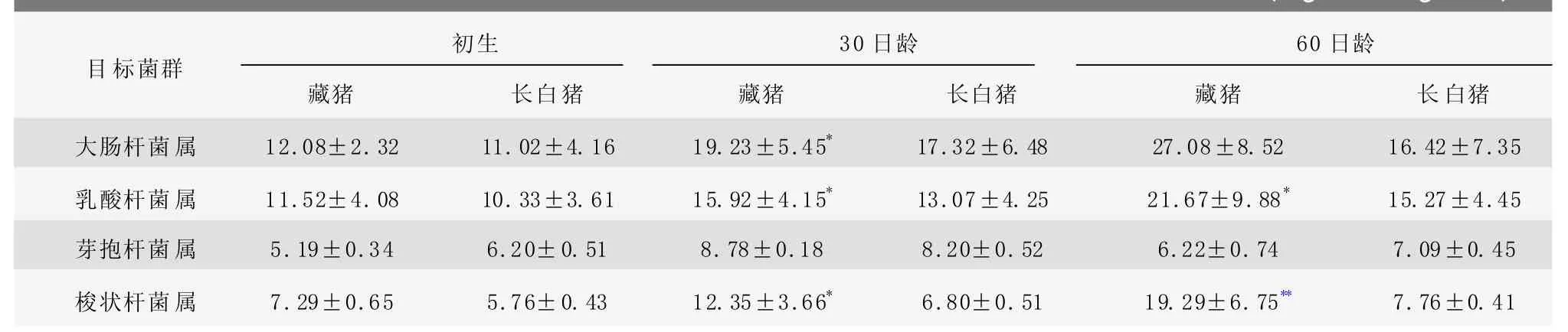
表5 常规组肠道内容物样品中几种菌群数量 (log拷贝数//gg样品)
表5结果显示,在出生时,藏猪、长白猪肠道内四种细菌的数量差异不显著。随着生长发育,断奶后藏猪肠道内大肠杆菌、乳酸杆菌显著多于长白猪(<0.05);藏猪肠道内梭状杆菌明显多于长白猪,30日龄时差异显著(<0.05),60日龄时差异极显著(<0.01);芽孢杆菌属两个猪种之间无明显差异。
2.3添加粗饲料后肠道微生物菌群的变化
表6结果显示,(1)添加粗饲料后,藏猪肠道内梭状杆菌、芽孢杆菌数量明显增加,与长白猪肠道内梭状杆菌、芽孢杆菌相比,差异极显著(<0.01);(2)添加粗饲料后,长白猪乳酸杆菌数量增加显著,而藏猪肠道内乳酸杆菌数量下降,差异显著(<0.05);(3)添加粗饲料后,两个猪种之间的大肠杆菌数量无明显差别。
3 小结与讨论
在哺乳动物中,肠道微生物在营养物质的摄取、上皮细胞的生长发育、免疫方面起着重要作用。猪的肠道中寄存着大量的微生物,复杂的肠道微生物区系对于肠道健康有着重要贡献[1]。近年来,随着分子生物学技术的发展,人们越发了解动物的肠道微生物群落,肠道微生物不仅是动物的一个重要代谢“器官”,而且也是动物自身重要的免疫“器官”,研究表明肠道有益微生物区系的建立对于避免仔猪腹泻和刺激肠道的生长、发育具有重要作用[2]。仔猪从刚出生到断奶后,直至体成熟,其肠道微生物的组成受到日粮、环境及宿主等因素共同影响,形成了一个相对稳定的多样化的微生物生态环境[3]。肠道微生物与宿主在漫长的协同进化过程中,相互选择并形成了一个相互依赖和相互制约的共生生态系统,肠道微生物是肠道生态系统的组成成员,并与宿主构成了一个统一的整体[4]。然而,肠道微生物区系的建立是一个非常复杂的过程,受品种、环境、发育阶段、消化道各部位组织结构和生理特性不同的影响,导致体内微生物菌落的数量、组成有所差异[5]。因此研究肠道微生物菌群的发育变化过程对于研究猪种的消化能力形成、以及对于粗饲料分解、营养成分吸收等具有重要意义。
我国的地方猪种普遍有耐粗饲的特性,肖文萍等[6]采用PCR/DGGE技术和16SrDNA序列分析方法,研究了普通猪、藏猪的肠道细菌优势菌群结构,发现猪肠道微生物区系的品种间差异大于品种内差异[6]。本研究结果显示:在出生时,藏猪、长白猪肠道内四种细菌的数量差异不显著;随着生长发育,断奶后藏猪大肠杆菌、乳酸杆菌数量显著多于长白猪(<0.05),藏猪梭状杆菌极显著地多于长白猪(<0.01),芽孢杆菌属两个猪种之间无明显差异。在添加粗饲料后,藏猪肠道内梭状杆菌、芽孢杆菌数量明显增加,与长白猪肠道内梭状杆菌、芽孢杆菌相比,差异极显著(<0.01);(2)添加粗饲料后,长白猪乳酸杆菌数量增加显著,藏猪肠道内乳酸杆菌数量降低,两者之间差异显著(<0.05);(3)两个猪种之间的大肠杆菌无明显差异。这些结果说明:不同品种的仔猪之间,肠道优势菌群呈现较大差异,肠道微生物群落差异与品种有直接相关。随着生猪日龄的增长,猪断奶及其对饲料(如增加粗饲料)与环境的适应,各品种猪粪样中微生物菌群发生了很大的变化,这些结果与杨柳等对地方品种荣昌猪[7]的研究结果类似。
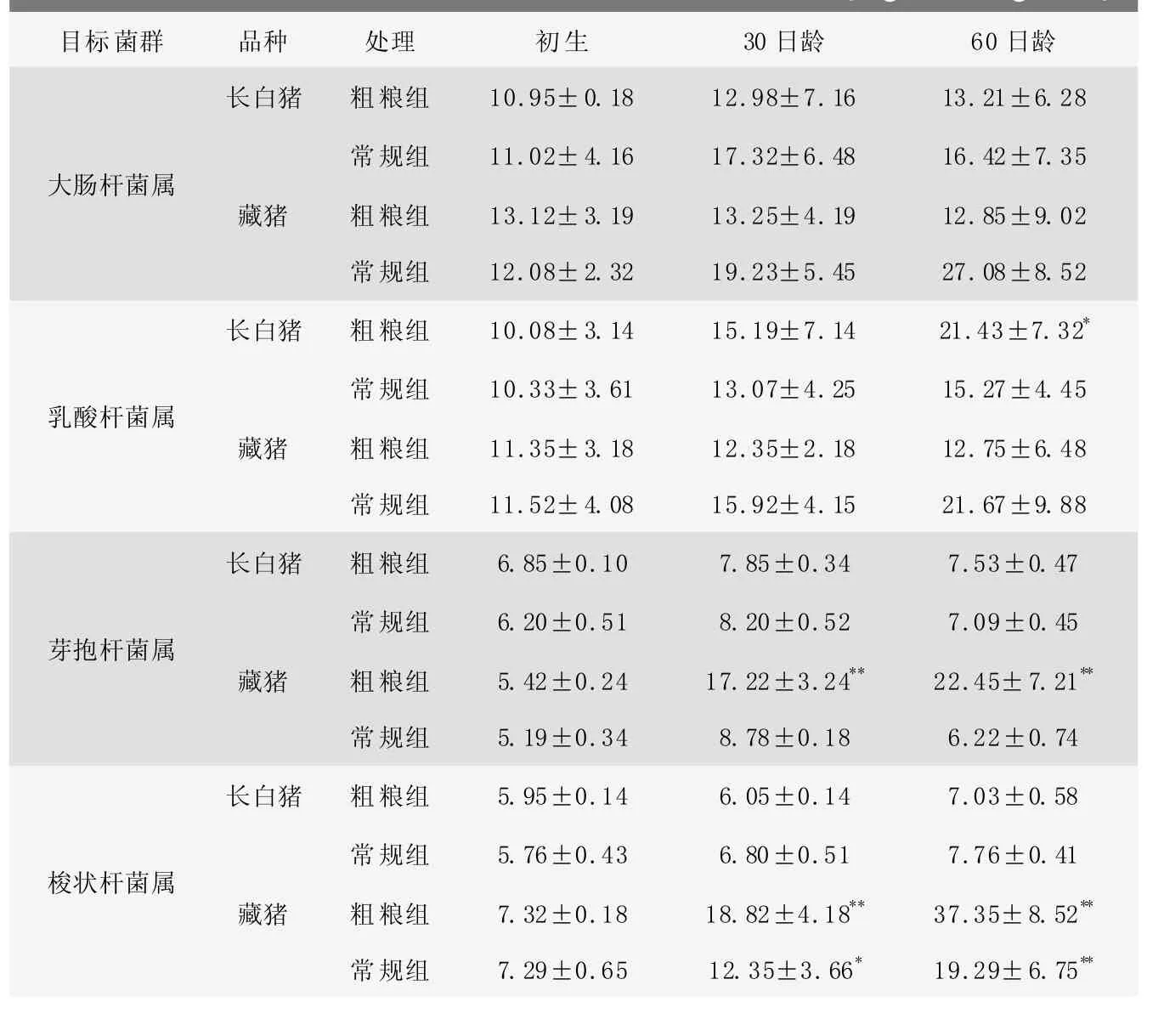
表6 常规组、粗粮组肠道内容物样品中几种菌群数量 (log拷贝数//gg样品)
4 结论
本研究结果表明,在出生时,藏猪、长白猪肠道内四种细菌的数量差异不显著;随着生长发育,断奶后藏猪肠道内大肠杆菌、乳酸杆菌数量显著多于长白猪(<0.05);梭状杆菌数量极显著多于长白猪(<0.01);芽孢杆菌属两个猪种之间无明显差异。添加粗饲料后,藏猪肠道内梭状杆菌、芽孢杆菌数量极显著增加(<0.01);长白猪肠道内乳酸杆菌数量显著增加(<0.05),两个猪种之间大肠杆菌无明显差异。可见,藏猪的耐粗性状与肠道内梭状杆菌、芽孢杆菌增加相关。
参考文献
[1]Mai V,Draganov PV.Recent advances and remaining gaps in our knowledge of associations between gut microbiota and human health[J].World J Gastroenterol,2009,15(1):81-85.
[2]Danielsen M,Hornshoj H,Siggers R H,et al.Effeets of baeterial colonization on the Porcine intestinal proteome[J].J Proteome Res,2007,6(7):2596-2604.
[3]Leser TD,Lindeerona RH,Jensen TK,et al.Changes in baeterial community structure in the colon of pigs fed different experimental diets and after infection with Brachyspira hyodysenteriae[J].APP Environ Microbiol,2000,66(8):3290-3296.
[4]Konstantinov SR,Awati AA,Williams BA,et al.Post -natal development of the porcine microbiota composition and activities[J].Environ Microbiol,2006,8(7):1191-1199.
[5]Ryo Inoue TT.Development of the intestinal microbita in the Piglets[J].J Gen APP Microbiol,2005,(51):257-265.
[6]肖文萍,刘海艳,赵海波,等.藏猪食草机理的研究——藏猪肠道微生物的多样性分析[J].中国兽医学报,2013,33(3):472-476.
[7]杨柳,张邑帆,郑华,等.荣昌、长白、杜洛克猪肠道微生物ERIC -PCRDGGE指纹图谱比较分析[J].家畜生态学报,2011,32(5):21-25.
作者简介:李江凌(1972-),女,研究员,从事动物遗传学研究,E-mail:yujiang1465@126.com
基金项目:四川省应用基础项目(2013JY0112);四川省财政基础科研项目;四川省生猪创新团队项目
收稿日期:2016-01-20
中图分类号:S828;S813.9
文献标识码:B
文章编号:1673-4645(2016)03-0061-04
