Protein inhibitor of activated STAT 4(PIAS4)regulates liver fi brosis through modulating SMAD3 activity
2016-04-18HuihuiXuZhiwenFanWenfangTianYongXu
Huihui Xu,Zhiwen Fan,Wenfang Tian,Yong Xu
Key Laboratory of Cardiovascular Disease and Molecular Intervention,Department of Pathophysiology,Nanjing Medical University,Nanjing,Jiangsu 211166,China.
Protein inhibitor of activated STAT 4(PIAS4)regulates liver fi brosis through modulating SMAD3 activity
Huihui XuΔ,Zhiwen FanΔ,Wenfang Tian,Yong Xu✉
Key Laboratory of Cardiovascular Disease and Molecular Intervention,Department of Pathophysiology,Nanjing Medical University,Nanjing,Jiangsu 211166,China.
Excessive fi brogenesis disrupts normal liver structure,impairs liver function,and precipitates the development of cirrhosis,an irreversible end-stage liver disease.A host of factors including nutrition surplus contribute to liver fi brosis but the underlying mechanism is not fully understood.In the present study,we investigated the involvement of protein inhibitor for activated stat 4(PIAS4)in liver fi brosis in a mouse model of non-alcoholic steatohepatitis (NASH).We report that PIAS4 silencing using short hairpin RNA(shRNA)attenuated high-fat high-carbohydrate (HFHC)diet induced liver f i brosis in mice.Quantitative PCR and Western blotting analyses conf i rmed that PIAS4 knockdown downregulated a panel of pro-f i brogenic genes including type I and type III collagens,smooth muscle actin,and tissue inhibitors of metalloproteinase.Mechanistically,PIAS4 silencing blocked the recruitment of SMAD3,a potent pro-f i brogenic transcription factor,to the promoter regions of pro-f i brogenic genes and dampened SMAD3 acetylation likely by upregulating SIRT1 expression.In conclusion,PIAS4 may contribute to liver f i brosis by modulating SIRT1-dependent SMAD3 acetylation.
liver f i brosis,PIAS4,SMAD3,acetylation,transcriptional regulation
Introduction
Non-alcoholic steatohepatitis or NASH has become a signif i cant health threat in a growing number of nations accompanying the global pandemic of obesity and type 2 diabetes[1].Liver f i brosis is one of the many complications associated with NASH[2].In response to nutrition surplus,several different types of cells including hepatic stellate cells and portal f i broblast cells trans-differentiate into pro-f i brogenic myof i broblasts and accelerate the production and deposition of extracellular matrix(ECM)proteins[3-4].Although considered a host defense mechanism,excessive fi brogenesis can disrupt normal liver structure and interfere with liver functionalities precipitating the development of end-stage liver diseases such as cirrhosis and hepatocellular carcinoma[5].The mechanism accounting for liver fi brosis during NASH pathogenesis is not fully appreciated.
Inside the ECM-producing cells, fi brogenesis is dictated by a network of growth factors,cytokines, and transcription factors[6].Transforming growth factor (TGF-β)is by far the most extensively studied profi brogenic factor in the liver,signaling primarily through the SMAD family of transcription factors[7]. Upon binding to its receptor,TGF-β triggers thephosphorylation and nuclear translocation of SMAD3. SMAD3 in turn binds to the promoter regions of profi brogenic genes(e.g.,type I collagen)and activates transcription.The ability of SMAD3 to promote profi brogenic transcription is in part impacted by its posttranslational modi fi cation status.In addition to phosphorylation,SMAD3 can also be acetylated in its lysine residues;acetylation of SMAD3 by the histone acetyltransferase p300/CBP enhances its activity[8].In contrast,deacetylation of SMAD3 by the lysine deacetylase SIRT1 dampens its activity[9].
Previously we have shown that protein inhibitor of activated STAT 4(PIAS4)downregulates SIRT1 expression at the transcriptional level in response to hypoxia in cancer cells[10-11].Therefore,we hypothesized that PIAS4 could potentially contribute to liver fi brosis by modulating SIRT1-dependent SMAD3(de) acetylation.Our data as summarized in this report support this hypothesis and indicate that targeting PIAS4 may provide novel therapeutic solutions against NASH-induced liver fi brosis.
Materials and methods
Animals
All animal protocols were approved by the NJMU Intramural Ethics Committee on Animal Studies.To induce steatohepatitis,8 week-old male C57/BL6 mice were fed a high fat high carbohydrate(HFHC)diet (D12492,Research Diets)for 16 consecutive weeks[12]. To knock down PIAS4,the mice were injected via the tail vein with purif i ed lentiviral particles(1X109MOI) that carry short hairpin RNA(shRNA)targeting PIAS4 (5'-GTGCTGTACGGGAAGTACTT-3')or scrambled shRNA(SCR)every 10 days for the duration of the experiments.
Protein extraction and Western blotting assay
Tissue lysates were obtained as previously described[13].Western blot analyses were performed with anti-SIRT1(Santa Cruz Biotechnology,Santa Cruz,CA,USA),anti-type III collagen(Santa Cruz Biotechnology),anti-PIAS4(Sigma),anti-β-actin (Sigma),anti-acetyl lysine(Cell Signaling Tech),antitype I collagen(Rockland),anti-α-SMA(Abcam),and anti-SMAD3(Abcam)antibodies.
Chromatin immunoprecipitation(ChIP)
ChIP assays were performed essentially as described before[14]with anti-SMAD3 antibody(Abcam).Precipitated genomic DNAwas amplif i ed by real-time PCR with primers as previously described[3,11,15].
Histology
Histological analyses were performed essentially as described before[11,13].Brief l y,paraff i n sections were stained with picrosirius red(Sigma)or Masson's trichrome(Sigma)according to standard procedures. Pictures were taken using an Olympus IX-70 microscope.
Statistical analysis
Data are presented as mean±SD.For experiments concerning multiple groups,one-way ANOVA with post-hoc Scheffe analyses were performed to evaluate the differences.The differences between two(control and experimental)groups were determined by twosided,unpaired Student's t-test.P values smaller than 0.05 are considered signif i cant.For the in vivo experiments,specif i c P values are spelled out.
Results
PIAS4 knockdown alleviates liver f i brosis in mice
Wef i rst examinedthe effect ofPIAS4 onliver f i brosis in vivo.To induceliver f i brosis,C57/BL6 micewere fed with a HFHC diet for 16 weeks[12].Picrosirius red(Fig. 1A)and Masson's trichrome(Fig.1B)staining revealed extensive interstitial f i brosis in the livers of mice fed on the HFHC diet compared to the mice on a control (chow)diet.PIAS4 knockdown was achieved via lentivirus-mediated delivery of shRNA injected through the tail vein.Western blotting analysis showed that compared to mice injected with control shRNA(SCR), PIAS4-specif i c shRNA(shPias4)signif i cantly downregulated PIAS4 levels in the liver(Fig.2B).Histological measurements showed that PIAS4 silencing largely abrogated HFHC diet induced liver f i brosis (Fig.1A and Fig.1B).
PIAS4 depletion downregulates expression of pro-f i brogenic genes
Next,we examined the effects of PIAS4 depletion on the expression of pro-f i brogenic genes in the liver. Quantitative PCR analyses showed that the HFHC diet stimulated the synthesis of a panel of pro-f i brogenic genes,including type I collagen(col1a1 and co1a2), type III collagen(col3a1),alpha smooth muscle actin (acta2),tissue inhibitors of matrix metalloproteinase (timp1 and timp3),lysyl oxidase(lox),and integrin subunit alpha 1(itga1).PIAS4 depletion,to varying extents,downregulated all the pro-f i brogenic genes examined here(Fig.2A).Western blotting experiments conf i rmed the observation that PIAS4 depletion systemically downregulated the induction of pro-f i brogenicgene expression by the HFHC diet(Fig.2B).Taken together,we were able to conclude that PIAS4 might be essential for liver f i brosis in an HFHC diet-induced model of NASH.

Fig.1 PIAS4 knockdown alleviates liver f i brosis in mice.C57/BL6 mice were fed on a high fat high carbohydrate(HFHC)-diet or a chow diet for 16 weeks.Lentivirus carrying either PIAS4 targeting shRNA or a control shRNAwas injected weekly via the tail vein.Picrosirius red(A) and Masson's trichrome(B)stainings were performed as described in Methods.Quantif i cation was carried out using Image Pro.N=5 mice for each group.Data are presented as mean±S.D.Scale bar,50 mm.
PIAS4 modulates SMAD3 activity by inf l uencing SIRT1-dependent deacetylation
SMAD3 is one of the most potent pro-f i brogenic transcription factors.We hypothesized that PIAS4 could modulate SMAD3 activity in the liver.ChIP assay showed that binding of SMAD3 to its target genes, including col1a1,col1a2,and acta2,was signif i cantly upregulated in the livers of mice fed on the HFHC diet (Fig.3A).On the contrary,PIAS4 silencing markedly dampened the occupancies of SMAD3 on its target promoters.
It has been documented that SIRT1 suppresses SMAD3 activity by promoting its deacetylation[9].As shown in Fig.2B,HFHC diet feeding caused a decrease in SIRT1 expression,consistent with an increase in SMAD3 activity;PIAS4 knockdown,however,was able to normalize SIRT1 expression,again in keeping with suppressed SMAD3 activity.We then examined the acetylation status of SMAD3 in various settings. Immunoprecipitation combined with Western blotting showed that HFHC diet feeding resulted in a signif i cant upregulation of SMAD3 acetylation,consistent with increased SMAD3 binding activity and decreased SIRT1 expression,in the liver(Fig.3B).PIAS4 depletion,however,blocked the induction of SMAD3 acetylation,which was in agreement with restored SIRT1 expression and reduced SMAD3 binding ontarget promoters.Collectively,these data suggest that PIAS4 might contribute to liver f i brosis possibly by modulating SIRT1-dependentdeacetylation of SMAD3.
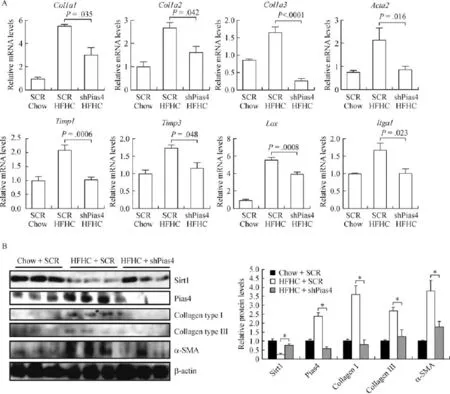
Fig.2 PIAS4 depletion downregulates expression of pro- fi brogenic genes.C57/BL6 mice were fed on an HFHC-diet or a chow diet for 16 weeks.Lentivirus carrying either PIAS4 targeting shRNA or a control shRNA was injected weekly via the tail vein.Expression levels of profi brogenic genes were examined by qPCR(A)and Western blotting assays(B).N=5 mice for each group.Data are presented as mean±S.D. *P<0.05.
Discussion
NASH serves as an intermediate disease state bridging the reversible and manageable steatosis and the irreversible and refractory cirrhosis[1].Liver f i brosis is a hallmark event in the pathogenesis of NASH,the ineffective intervention of which may precipitate the development end-stage liver diseases and signif i cantly dim the chance of patient survival.We report here that PIAS4 may play a critical role in liver f i brosis by modulating SMAD3 activity,likely through SIRT1-dependent deacetylation.Several previous investigations have implicated PIAS1 as a modulator of the TGF-β pathway although the conclusions seem to be contradictory.For instance,the Kurabayashi group has reported that PIAS1 is essential for TGF-β induced α-SMA trans-activation in smooth muscle cells by SUMOylating KLF4[16].In contrast,Netherton and Bonni demonstrated that PIAS1 represses TGF-β initiated mesenchymal cell differentiation by SUMOylating SnoN[17].Our data suggest that PIAS4 modulates TGF-β signaling in an indirect manner,namely,through SIRT1-dependent deacetylation of SMAD3.However, the possibility that PIAS4 could directly interact with and SUMOylate SMAD3 cannot be excluded especially in light of the f i nding that SMAD4,the common SMAD protein required for TGF-β signaling,have been found to be a direct substrate for SUMOylation[18].Inaddition,it remains unclear whether other members of the PIAS family could play a non-redundant role in regulating liver f i brosis.Starkel et al.have shown that PIAS3 expression was progressively increased during the development of liver f i brosis/cirrhosis in patients infected with hepatitis C virus(HCV),suggesting that PIAS3 may also play a precipitating role in this process[19].When further conf i rmed,our data add support to the argument of using a pan-PIAS inhibitor to stall or reverse f i brogenesis in the liver.

Fig.3 PIAS4 modulates SMAD3 activity by inf l uencing SIRT1-dependent deacetylation.C57/BL6 mice were fed on an HFHC-diet or a chow diet for 16 weeks.Lentivirus carrying either PIAS4 targeting shRNA or a control shRNAwas injected weekly via the tail vein.(A)ChIP assay was performed using liver homogenates with anti-SMAD3 antibody.Precipitated DNA was amplif i ed using primers surrounding the indicated gene promoters.(B)Immunoprecipitation was performed with anti-SMAD3 using liver homogenates.Western blotting was performed with anti-SMAD3 or anti-acetyl lysine.n=3 mice for each group.Data are presented as mean±S.D.*P<0.05.
We show here that PIAS4 promotes liver f i brosis likely through SIRT1-dependent SMAD3 deacetylation. In fact,several alternative scenarios exist to interpret the data.First,PIAS4 has been known to modulate cellular response to hypoxia[10-11,20],which by itself is a risk factor for NASH and a promoter of liver f i brosis[21]. Second,liver f i brosis in the context of NASH often occurs as a result of excessive hepatic inf l ammation. PIAS4 can directly SUMOylate and thus activate NF-kB,the master regulator of cellular inf l ammation[22]. Therefore,our observation that PIAS4 knockdown attenuated liver f i brosis could be secondary to reduced hepatic inf l ammation as a result of NF-kB deactivation. Finally,we used a lentivirus delivery system that did not differentiate the liver from other organs or cells in the circulation.It is possible that PIAS4 might inf l uence liver f i brosis by regulating circulating myeloid cells(e. g.,macrophages),which are considered a driving force of liver f i brosis[23].These remaining issues will have to be sorted out by future investigations.
In summary,we provide evidence that PIAS4 knockdown in a mouse model of NASH effectively attenuated liver f i brosis.Therefore,PIAS4 could become an attractive target for the development of novel therapeutic strategies to prevent excessive liver f i brogenesis.
Acknowledgements
This work was supported by the Natural ScienceFoundation of China(No.81500441).YX is a Fellow at the Collaborative Innovation Center for Cardiovascular Disease Translation Research.
[1] Marchesini G,Petta S,Dalle Grave R.Diet,weight loss,and liver health in nonalcoholic fatty liver disease:Pathophysiology,evidence,and practice[J].Hepatology,2015.
[2]Argo CK,Northup PG,Al-Osaimi AM,et al.Systematic review of risk factors for f i brosis progression in non-alcoholic steatohepatitis[J].J Hepatol,2009,51(2):371–379.
[3] Tian W,Hao C,Fan Z,et al.Myocardin related transcription factor A programs epigenetic activation of hepatic stellate cells [J].J Hepatol,2015,62(1):165–174.
[4] Fan Z,Hao C,Li M,et al.MKL1 is an epigenetic modulator of TGF-β induced f i brogenesis[J].Biochim Biophys Acta,2015, 1849(9):1219–1228.
[5] Tian W,Xu Y.Decoding liver injury:A regulatory role for histone modif i cations[J].Int J Biochem Cell Biol,2015,67: 188–193.
[6] Hernandez-Gea V,Friedman SL.Pathogenesis of liver f i brosis [J].Annu Rev Pathol,2011,6:425–456.
[7] Moustakas A,Souchelnytskyi S,Heldin CH.Smad regulation in TGF-beta signal transduction[J].J Cell Sci,2001,114(Pt 24): 4359–4369.
[8] InoueY,Itoh Y,Abe K,et al.Smad3 is acetylated byp300/CBP to regulate its transactivation activity[J].Oncogene,2007,26 (4):500–508.
[9] Li J,Qu X,Ricardo SD,et al.Resveratrol inhibits renal f i brosis in the obstructed kidney:potential role in deacetylation of Smad3[J].Am J Pathol,2010,177(3):1065–1071.
[10]Sun L,Li H,Chen J,et al.PIASy mediates hypoxia-induced SIRT1 transcriptional repression and epithelial-to-mesenchymal transition in ovarian cancer cells[J].J Cell Sci,2013,126(Pt 17):3939–3947.
[11]Sun L,Li H,Chen J,et al.A SUMOylation-dependent pathway regulates SIRT1 transcription and lung cancer metastasis[J].J Natl Cancer Inst,2013,105(12):887–898.
[12]Kohli R,Kirby M,Xanthakos SA,et al.High-fructose,medium chain trans fat diet induces liver f i brosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis[J].Hepatology,2010,52(3): 934–944.
[13]Tian W,Xu H,Fang F,et al.Brahma-related gene 1 bridges epigenetic regulation of proinf l ammatory cytokine production to steatohepatitis in mice[J].Hepatology,2013,58(2):576–588.
[14]Fang F,Chen D,Yu L,et al.Proinf l ammatory stimuli engage Brahma related gene 1 and Brahma in endothelial injury[J]. Circ Res,2013,113(8):986–996.
[15]Kim M,Yi SA,Lee H,et al.Reversine induces multipotency of lineage-committed cells through epigenetic silencing of miR-133a[J].Biochem Biophys Res Commun,2014,445(1):255–262.
[16]Kawai-Kowase K,Ohshima T,Matsui H,et al.PIAS1 mediates TGFbeta-induced SM alpha-actin gene expression through inhibition of KLF4 function-expression by protein sumoylation [J].Arterioscler Thromb Vasc Biol,2009,29(1):99–106.
[17]Netherton SJ,Bonni S.Suppression of TGFβ-induced epithelial-mesenchymal transition like phenotype by a PIAS1 regulated sumoylation pathway in NMuMG epithelial cells[J]. PLoS One,2010,5(11):e13971.
[18]Lin X,Liang M,Liang YY,et al.Activation of transforming growth factor-beta signaling by SUMO-1 modif i cation of tumor suppressor Smad4/DPC4[J].J Biol Chem,2003,278(21): 18714–18719.
[19]Starkel P,Saeger CD,Leclercq I,Horsmans Y.Role of signal transducer and activator of transcription 3 in liver f i brosis progression in chronic hepatitis C-infected patients[J].Laboratory investigation;a journal of technical methods and pathology.2007;87(2):173–81.Epub 2007/02/24.
[20]Cai Q,Verma SC,Kumar P,et al.Hypoxia inactivates the VHL tumor suppressor through PIASy-mediated SUMO modif i cation[J].PLoS One,2010,5(3):e9720.
[21]Byrne CD.Hypoxia and non-alcoholic fatty liver disease[J]. Clin Sci(Lond),2010,118(6):397–400.
[22]Mabb AM,Wuerzberger-Davis SM,Miyamoto S.PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress[J].Nat Cell Biol,2006,8(9):986–993.
[23]Lech M,Anders HJ.Macrophages and f i brosis:How resident and inf i ltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair[J].Biochim Biophys Acta,2013,1832 (7):989–997.
ΔThese authors contributed equally to this work.
✉Corresponding author:Yong Xu,PhD,Nanjing Medical University,101 Longmian Avenue,Jiangning District,Nanjing,Jiangsu 211166,China.Email:yjxu@njmu.edu.cn,Tel/Fax:+86-25-86862888/+86-25-86862888
©2016 by the Journal of Biomedical Research.All rights reserved
Received 9 April 2016,Revised 29 June 2016,Accepted 11 July 2016,Epub 1 August 2016
R657.31,Document code:A
The authors reported no conf l ict of interests.
10.7555/JBR.30.20160049
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Pathology and molecular characterization of recent Leucocytozoon caulleryi cases in layer f l ocks
- Modified methods for isolation of pancreatic stellate cells from human and rodent pancreas
- Retrograde traff i cking of VMAT2 and its role in protein stability in non-neuronal cells
- Chronic intermittent hypoxia induces cardiac inf l ammation and dysfunction in a rat obstructive sleep apnea model
- Assessment of atrial electromechanical interval using echocardiography after catheter ablation in patients with persistent atrial f i brillation
- Elevated thyroid stimulating hormone levels are associated with metabolic syndrome in a Chinese community-based population of euthyroid people aged 40 years and older