磷氯钡石[Ba5(PO4)3Cl]在水溶液中的溶解作用及溶度积
2016-04-17朱宗强魏彩春朱义年贺利娜
林 局,朱宗强,潘 艳,魏彩春,3,朱义年,贺利娜
(1.桂林理工大学a.岩溶地区水污染控制与用水安全保障协同创新中心;b.广西环境污染控制理论与技术重点实验室,广西桂林 541004;2.广西壮族自治区环境监测中心站,南宁 530028;3.广西大学轻工与食品工程学院,南宁 530004)
磷氯钡石[Ba5(PO4)3Cl]在水溶液中的溶解作用及溶度积
林 局1,朱宗强1,潘 艳2,魏彩春1,3,朱义年1,贺利娜1
(1.桂林理工大学a.岩溶地区水污染控制与用水安全保障协同创新中心;b.广西环境污染控制理论与技术重点实验室,广西桂林 541004;2.广西壮族自治区环境监测中心站,南宁 530028;3.广西大学轻工与食品工程学院,南宁 530004)
合成和表征了磷氯钡石[Ba5(PO4)3Cl]固体,通过溶解实验研究了其在水溶液中的溶解作用。结果表明:溶液的pH值和组分随时间的变化与初始pH值密切相关。溶解过程包括了质子化作用,Ba2+、PO和Cl-离子的释放、络合作用,重结晶作用等。在25℃和初始pH=2.00、5.60、9.00的条件下,磷氯钡石[Ba5(PO4)3Cl]的溶度积(Ksp)分别为10-42.85、10-43.44和10-43.34,相应的吉布斯生成自由能ΔGfo分别为-6 235.82、-6 239.16和 -6 238.58 kJ/mol。一些常见的氯磷灰石矿物在水中的稳定性顺序为Pb5(PO4)3Cl>Cd5(PO4)3Cl>Ca5(PO4)3Cl>Ba5(PO4)3Cl>Sr5(PO4)3Cl>Zn5(PO4)3Cl。
磷氯钡石;溶解作用;溶度积;吉布斯生成自由能
降低生物有效性与生物毒性是重金属污染土壤修复的重要手段之一,其中含磷材料可作为铅的稳定修复剂而得到广泛利用。含磷材料可通过沉淀、离子交换等作用与铅离子形成稳定的磷氯铅矿类物质[Pb5(PO4)3Cl][1]。磷氯铅矿属于磷灰石族,天然矿物于1748年首先发现于法国,其后在前苏联、英国、非洲、澳大利亚等地均有发现。1992年广西阳朔铅锌矿的氧化带中曾发现少量该矿物,2001年广西恭城县岛坪铅锌矿发现结晶非常好的磷氯铅矿[2]。磷氯铅矿物中的Ba、Ca和CO3是因矿石沉积过程中所产生的类质同象现象而进入到矿石的结构中[3]。磷氯铅矿中的Pb2+可以被Ba2+类质同象取代形成磷氯钡石[Ba5(PO4)3Cl]或[(Pb,Ba)5(PO4)3Cl]固溶体[4],从而影响磷氯铅矿在环境中的稳定性,但评价这些物质的形成和稳定性所需要的热力学数据目前报道较少。Khattech等[5]通过实验得出 25℃时磷氯钡石[Ba5(PO4)3Cl]的溶度积为10-110.69,而Jemal[6]通过理论计算给出的 Ba5(PO4)3Cl的溶度积为10-52.5,者相差甚大。所以,有必要对其热力学数据重新进行测定计算。
笔者首先合成了磷氯钡石 [Ba5(PO4)3Cl]固体,并利用X射线衍射 (XRD)、傅里叶红外光谱 (FT-IR)和扫描电子显微镜 (SEM)对其进行表征;然后通过溶解实验,研究磷氯钡石在不同pH值条件下的溶解作用过程,依据分析数据计算得出了磷氯钡石在25℃下的溶度积 (Ksp)和吉布斯生成自由能。
1 实验部分
1.1 合成实验
采用共沉淀法按照化学式5Ba2++3PO34-+Cl-=Ba5(PO4)3Cl合成磷氯钡石[Ba5(PO4)3Cl]固体。首先,用超纯水将上海国药集团化学试剂有限公司生产的NaCl、Na3PO4和Ba(C2H3O2)2分别配制成 0.5 mol/L的 NaCl溶液、0.5 mol/L的Na3PO4溶液、0.5 mol/L的 Ba(C2H3O2)2溶液。然后在一个250 mL的碘量瓶中,依次加入100 mL 0.5 mol/L的Ba(C2H3O2)2溶液、20 mL 0.5 mol/L的NaCl溶液和60 mL 0.5 mol/L的Na3PO4溶液。盖好瓶盖后置于50℃的恒温水浴槽中,并定期均匀摇动。经过22 d的反应后进行固液分离,用乙醇和超纯水洗净固相沉淀物,并将其放在90℃恒温鼓风干燥箱中连续干燥5 d,在沉淀物的温度自然降到室温后装入棕色小瓶里备用。称取1 g固体用优级纯的浓HNO3消解后,用原子吸收光谱仪(Perkin-Elmer AAnalyst 700)测定液相中Ba的浓度,用离子色谱仪(Dionex ICS1000)测定液相中P和Cl的浓度,计算合成固体的组成。利用X射线衍射仪(XRD,X'Pert PRO)、傅里叶红外光谱仪(470 FT-IR)和扫描电子显微镜(SEM,JSM-6380LV;配置X射线能谱仪EDS,型号IE 350)对固相沉淀物进行表征,分析其晶体结构、结晶状况和形貌等特征。
1.2 溶解实验
分别称取1.5 g固体置于3个250 mL的聚乙烯瓶中。然后,在每个瓶中分别加入100 mL初始pH值为2.00的HNO3溶液、初始pH值5.60超纯水和初始pH值9.00的NaOH溶液。将这3个聚乙烯瓶密封后置于振荡恒温水浴锅中溶解,温度设为(25±1)℃,振荡频率设为50 r/min。在实验过程中,对溶解的固体上清液进行定时采样,分析溶液中组分以实时监控液相和固相的变化。随溶解时间不同共采集19次(1、3、6、12、24、48、72、120、240、360、480、720、960、1 080、1 200、1 440、1 800、2 880和3 600 h)。每次取样前,先测定溶解的固体上清液pH,然后采集6 mL的上清液并用0.20μm的针筒过滤器过滤到小瓶中。分别用石墨炉原子吸收光谱法测定液相中Ba的浓度,用离子色谱法测定P和Cl的浓度。溶解3 600 h后,从聚乙烯瓶中取出固体并用超纯水和乙醇洗净,后将其放置在90℃的电热恒温箱中连续干燥5 d。用X射线衍射仪、红外光谱仪和扫描电子显微镜分析溶解后固体的晶体结构、结晶状况和形貌等特征。
2 结果与讨论
2.1 固相表征
合成的磷氯钡石[Ba5(PO4)3Cl]固体的实际组分取决于混合溶液的nBa/nP和nBa/nCl值。化学分析结果表明,本实验中所合成的磷氯钡石的nBa/nP和nBa/nCl值分别为1.67和5.00,与其理论值一致。
XRD分析结果表明,合成的固体为磷氯钡石[Ba5(PO4)3Cl],与 JCPDS标准卡号01-070-2318(磷氯钡石Alforsite)一致,为六方晶系,属P63/m空间群,晶胞参数为a=b=1.028 4 nm,c =0.765 1 nm。而与羟基磷灰石(JCPDS 00-024-0033)衍射峰的位置相比,磷氯钡石衍射峰的位置出现非常显著的向高角度偏移(图1)。在25℃和初始pH=2条件下溶解3 600 h后,磷氯钡石固体的衍射峰及其位置未发生显著变化,表明晶体结构未发生改变,同时在溶解过程中也未形成新的固相物质。

图1 磷氯钡石[Ba5(PO4)3Cl]固体溶解前(A)、在25℃和初始pH=2.00条件下溶解3 600 h后(B)的XRD图谱Fig.1 X-ray diffraction(XRD)patterns of alforsite[Ba5(PO4)3Cl]before(A)and after(B)dissolution at25℃with an initial pH of 2.00 for 3 600 h
红外光谱分析结果表明,磷氯钡石固体样品在溶解前后的红外吸收光谱一致,晶体结构未发生改变(图2)。自由PO34-离子有4个基频频率,即对称伸缩振动ν1、弯曲振动ν2、反对称伸缩振动ν3和弯曲振动ν4[7]。样品中的磷酸根四面体在440~442 cm-1处有O—P—O的弯曲振动(ν2)吸收带,在935 cm-1处有P—O伸缩振动 (ν1)吸收带,在1 011~1 155 cm-1处有P—O的反对称伸缩振动(ν3)吸收带,在553~580 cm-1处有O—P—O弯曲振动(ν4)吸收带。磷氯钡石样品中吸附水的O—H伸缩振动吸收峰位于3 575 cm-1左右,O—H的受阻转动带则位于654 cm-1左右。在谱图中,都没有出现明显的CO和HPO的振动吸收带。
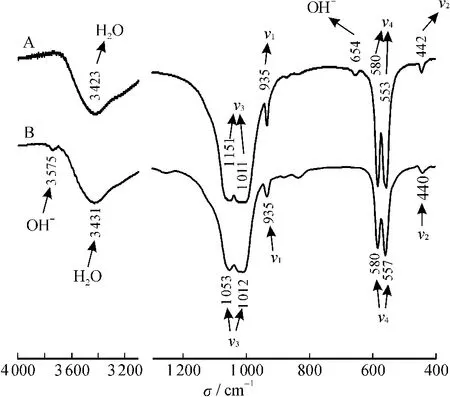
图2 磷氯钡石[Ba5(PO4)3Cl]固体溶解前(A)和在25℃和初始pH=2条件下溶解3 600 h后(B)的FT-IR图谱Fig.2 Fourier transform infrared(FT-IR)spectra of alforsite[Ba5(PO4)3Cl]before(A)and after(B)dissolution at25℃with an initial pH of2.00 for 3 600 h
晶体的形貌受外部生长环境条件的影响[8]。扫描电镜分析结果表明,磷氯钡石固体样品在溶解前后的结晶状况和形貌发生显著变化 (图3)。合成的磷氯钡石固体样品在溶解前呈现非常细小的针状集合体(<0.2μm),但在25℃和初始pH=2条件下溶解3 600 h后,晶体呈带锥面的六方柱,晶体颗粒显著变大,粒径可达100μm以上,表明磷氯钡石固体样品在溶解过程发生了较为强烈的重结晶作用。
2.2 溶液组分随时间的变化
磷氯钡石[Ba5(PO4)3Cl]固体在25℃和不同初始pH条件下溶解时,溶液的pH值和组分随时间的变化趋势与其初始pH值密切相关(图4)。
在初始pH=2条件下开始溶解时,溶液的pH值随溶解时间增加而从2.00逐渐升高至7.50~7.60,在溶解240 h后基本达到稳定状态;溶液中Ba的浓度在溶解开始后的1 h内浓度就已达到9.78 mmol/L,然后由于溶解与重结晶的共同作用,随溶解时间增加而在5.63~9.71 mmol/L范围内变动;溶液中P的浓度在溶解开始后的1 h内浓度就已达到1.27 mmol/L,然后随溶解时间增加而逐渐降低,溶解48 h后减小至0.08 mmol/L,在这之后基本达到稳定状态;溶液中Cl的浓度随溶解时间增加而升高,在溶解72 h后达到2.50 mmol/L,然后随溶解时间增加而在2.41~3.08 mmol/L范围内变动,在溶解2 160 h后达到稳定状态。
在初始pH=5.60和9.00条件下开始溶解时,溶液的pH值在溶解1 h内就从5.60和9.00迅速升高至9.05和9.21,然后缓慢降低至8.20~8.30左右,在溶解720 h后基本达到稳定状态;溶液中Ba的浓度随溶解时间增加而逐渐升高,溶解12~24 h后分别达到0.88和0.90 mmol/L,然后缓慢升高,在溶解1 080 h后基本达到稳定状态,为1.03~1.13和1.06~1.29 mmol/L;溶液中P的浓度随溶解时间增加而非常缓慢地升高,在溶解1 800 h时达到0.13 mmol/L,然后处于稳定状态;溶液中Cl的浓度随溶解时间增加而非常缓慢地升高,在溶解480~720 h后分别达到0.41和0.39 mmol/L,然后基本处于稳定状态。
2.3 溶解作用机制的分析

图3 磷氯钡石[Ba5(PO4)3Cl]固体溶解前(A)和在25℃和初始pH=2条件下溶解3 600 h后(B、C、D)的SEM图谱Fig.3 Field emission scanning electron microscopy(FE-SEM)images of alforsite[Ba5(PO4)3Cl]before(A)and after(B,C,D) dissolution at25℃with an initial pH of2.00 for 3 600 h
磷氯钡石[Ba5(PO4)3Cl]固体在25℃和不同初始pH条件下溶解时,溶液的pH值在溶解开始阶段均呈现升高,即H+被消耗而出现浓度降低,表明溶液中H+被吸附到磷氯钡石固体中PO四面体的带负电荷的氧离子上,导致固体表面上的PO转变为HPO,从而进一步促进了磷氯钡石在水中的溶解作用。要完整描述溶解过程中H+的消耗,就必须考虑固体的化学计量溶解、固体表面上H+与Ba2+之间的离子交换、H+吸附和解吸附等。磷灰石类矿物在水中的溶解过程与实验条件密切相关,尽管在文献中已经有许多溶解模型,但都仅仅考虑了磷灰石溶解过程的某些特定方面,而不具有普遍性[9]。
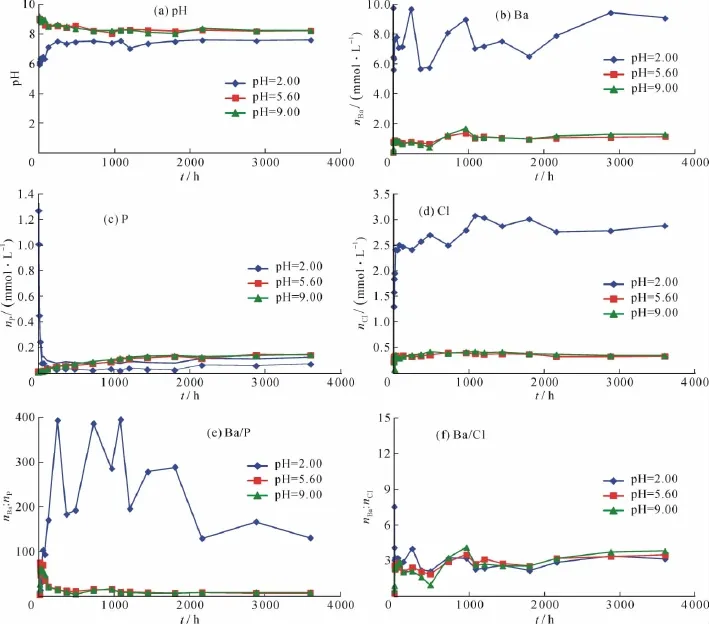
图4 Alforsite[Ba5(PO4)3Cl]在25℃和不同初始pH值下溶解过程中液相的变化Fig.4 Aqueous evolution during the dissolution of alforsite[Ba5(PO4)3Cl]at25℃ and different initial pHs
基于本实验以及前人的研究结果,笔者认为磷氯钡石在水溶液中溶解时,主要包括以下几个同时发生的作用过程:①H+从溶液到固-液界面的扩散;②H+被吸附到固相表面;③PO固相表面的存在形态发生转变;④Ba2+、PO和Cl-离子从固相表面释放进入溶液中;⑤Ba2+、PO和Cl-离子在溶液中发生络合作用;⑥Ba2+和PO被重新吸附到固相表面,发生重结晶作用;⑦达到稳定状态。
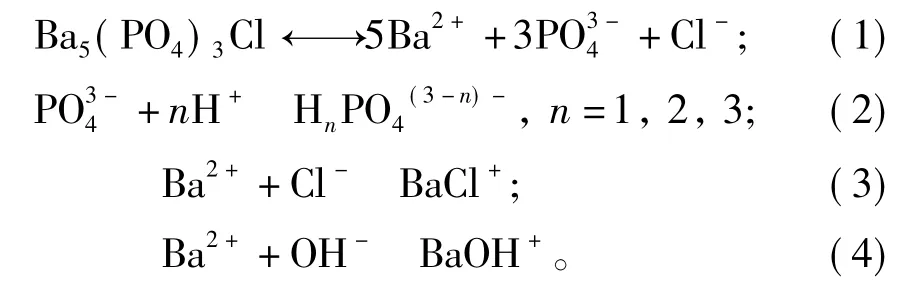
在反应过程①~④中,按照化学反应式(1),Ba2+、PO和Cl-离子按磷氯钡石[Ba5(PO4)3Cl]的摩尔比(nBa/nP=1.67、nBa/nCl=5)被同时溶解释放,从固相进入到溶液中。在作用过程⑤中,可能存在多种络合化学反应(2)、(3)和(4),从而导致溶液pH值的升高或降低。
在初始pH=2.00条件下,磷氯钡石溶解开始时溶液中磷酸盐的存在形态主次顺序为H2PO>,但在溶解120 h后变化为Ba和Cl的主要形态为游离态的Ba2+和Cl-离子。在初始pH= 5.60和9.00条件下,溶液中磷酸盐的存在形态主次顺序始终为Ba和Cl的主要形态为游离态的Ba2+和Cl-离子。
2.4 溶度积的计算
磷氯钡石[Ba5(PO4)3Cl]在水溶液中的溶解作用可以用方程式(1)表达,则Ba5(PO4)3Cl的离子活度积IAP为
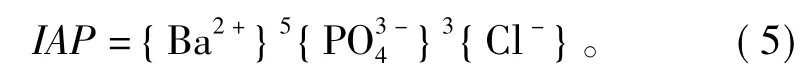
式中:{}内为组分的活度。
溶解平衡时,溶度积 Ksp=log IAP。首先用PHREEQC软件计算的活度。计算过程中,溶液中钡存在形态包括Ba2+、BaCl+和 BaOH+;磷的存在形态为。这些存在形态的基本物理化学性质参数主要来源于PHREEQC中自带的minteq.v4.dat热力学数据库[10]。然后利用式(5)计算出磷氯钡石[Ba5(PO4)3Cl]的离子活度积IAP,再以溶解达到平衡或稳定状态时(1 200、2 880和3 600 h)的IAP作为其溶度积Ksp(表1)。
磷氯钡石[Ba5(PO4)3Cl]溶解反应的自由能(ΔG)取决于其溶度积K:sp
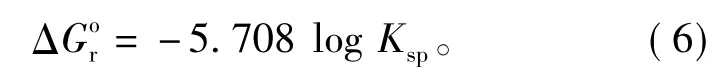
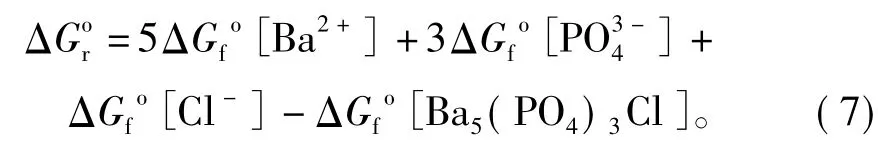
整理,得
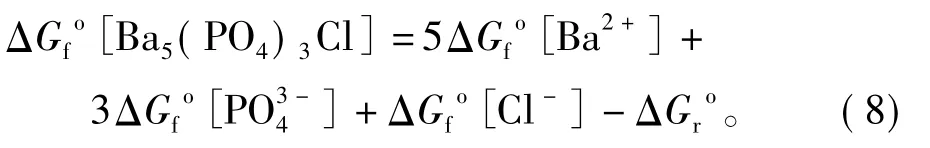
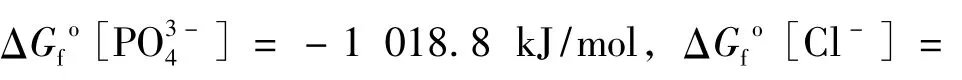
计算可知,在25℃和初始pH=2.00、5.60、9.00的条件下,磷氯钡石[Ba5(PO4)3Cl]的溶度积Ksp分别为 10-42.85(10-41.91~ 10-44.30)、10-43.44(10-43.20~10-43.73)和10-43.34(10-42.87~10-44.10),相应的吉布斯生成自由能ΔG[Ba5(PO4)3Cl]分别为-6 235.82、-6 239.16和-6 238.58 kJ/mol(表1)。结果显示,在不同初始pH实验条件下,所得到的磷氯钡石的溶度积和生成自由能大小基本一致。同时,磷氯钡石在水中的溶解能力要比前人的研究结果大得多。Khattech等[5]通过实验得出25℃时Ba5(PO4)3Cl的吉布斯生成自由能-6 623 kJ/mol,相应的溶度积为10-110.69;基于Khattech等[5]实验结果,Jemal[6]通过理论计算了Ba5(PO4)3Cl的吉布斯生成自由能-6 269.5 kJ/mol,相应的溶度积为10-52.5; Yoder等[12]在实验研究碳酸化磷氯钡石的溶解过程时,推断非碳酸化磷氯钡石的溶度积为10-51.5,吉布斯生成自由能为-6 285.16 kJ/mol。可见,这些文献所给出的结果都存在明显的偏差与局限性。同时,与文献中给出的其他氯磷灰石的Ksp比较可知,氯磷灰石的稳定性顺序一般为:Pb5(PO4)3Cl>Cd5(PO4)3Cl>Ca5(PO4)3Cl>Ba5(PO4)3Cl>Sr5(PO4)3Cl>Zn5(PO4)3Cl,它们的溶度积Ksp依次 为 10-84.4~-86.50[13-14]、10-63.1[4]、10-57.7[15]、10-41.91~44.10(本文结果)、10-41[6]和 10-38.1[4]。因此,在利用钡锶钙磷酸盐稳定化/固化含重金属废物时,对含Pb和Cd废物的处理效果较好,而对含Zn废物的处理效果较差;在利用钡锶钙磷酸盐进行重金属污染土壤修复时,可有效降低Pb和Cd的生物有效性,但提高了Zn的生物有效性。
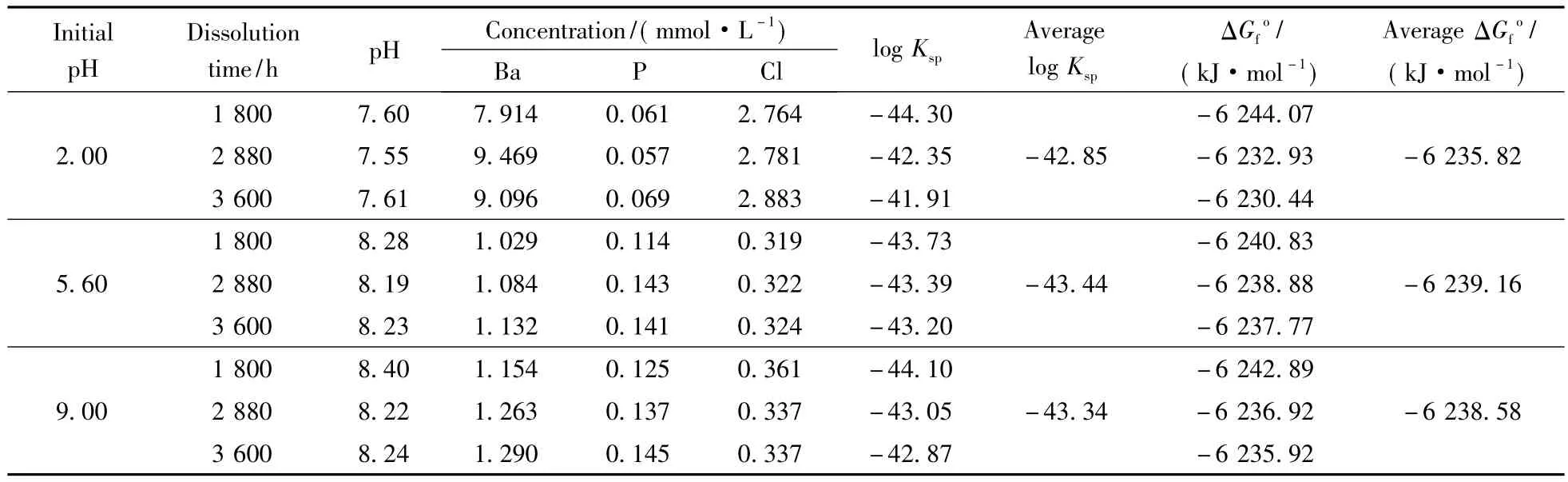
表1 Alforsite[Ba5(PO4)3Cl]的溶度积计算结果(25℃)Table 1 Solubility determination of alforsite[Ba5(PO4)3Cl](25℃)
3 结论
XRD、FT-IR和SEM的分析结果显示,磷氯钡石[Ba5(PO4)3Cl]固体在溶解前后的化学组成和结构没有发生显著变化,但在初始pH=2.00条件下溶解时,发生强烈的重结晶作用而使晶体颗粒的粒径显著增大。磷氯钡石固体在25℃和不同初始pH条件下溶解时,溶液的pH值和组分随时间的变化趋势与其初始pH值密切相关。磷氯钡石在水溶液中的溶解包括了H+在溶液中扩散并被吸附到固相表面,Ba2+、PO和Cl-离子从固相表面释放进入溶液中并发生络合作用,Ba2+和PO被重新吸附到固相表面而发生重结晶作用等几个同时发生的过程。在25℃和初始pH=2.00、5.60、9.00的条件下,磷氯钡石[Ba5(PO4)3Cl]的溶度积Ksp分别 为 10-42.85(10-41.91~ 10-44.30)、10-43.44(10-43.20~10-43.73)和10-43.34(10-42.87~10-44.10),相应的吉布斯生成自由能ΔG[Ba5(PO4)3Cl]分别为-6 235.82、-6 239.16和-6 238.58 kJ/mol。不同氯磷灰石在水溶液中的稳定性顺序为Pb5(PO4)3Cl> Cd5(PO4)3Cl> Ca5(PO4)3Cl>Ba5(PO4)3Cl>Sr5(PO4)3Cl>Zn5(PO4)3Cl。
[1]宋波,曾炜铨,陆素芬,等.含磷材料在铅污染土壤修复中的应用[J].环境工程学报,2015,9(12):5649-5658.
[2]马红艳,崔大安,秦作路,等.广西岛坪磷氯铅矿的谱学特征[J].矿物学报,2006,26(2):165-168.
[3]Botto IL,Barone V L,Castiglioni JL,et al.Characterization of a natural substituted pyromorphite[J].Journal of Materials Science,1997,32(24):6549-6553.
[4]Shevade A V,Erickson L,PierzynskiG,et al.Formation and stability of substituted pyromorphite:A molecular modeling study[J].Journalof Hazardous Substance Research,2001,3 (2):1-12.
[5]Khattech I,Lacout JL,Jemal M.Synthesis and thermochemistry of alkali earth phosphates.I.Standard enthalpies of formation of tribaritic phosphate and phosphobaritic chlorapatite[J].Annales De Chimie Science Des Matériaux,1996,21 (4):251-258.
[6]Jemal M.Thermochemistry and kinetics of the reactions of apatite phosphateswith acid solutions[C] //TadashiM.Application of Thermodynamics to Biological and Materials Science.Rijeka:InTech,2011:547-572.
[7]彭文世,刘高魁,柯丽琴.某些磷灰石矿物的红外吸收光谱[J].矿物学报,1986,6(1):26-35.
[8]阮青锋,邱志惠,秦晴,等.BaCl2·2H2O晶体聚集体形貌及结晶学规律[J].桂林理工大学学报,2014,34(3): 519-525.
[9]Dorozhkin SV.A review on the dissolution models of calcium apatites[J].Progress in Crystal Growth&Characterization of Materials,2002,44(1):45-61.
[10]Parkhurst D L,Appelo CA J.Description of inputand examples for PHREEQC version 3—A computer program for speciation,batch-reaction,one-dimensional transport,and inverse geochemical calculations[R].U.S.Department of the Interioy,U.S. Geological Survey.Denver:U.S. Gedogy Survey,2013.
[11]Stumm W,Morgan J J.Aquatic Chemistry,Chemical Equilibria and Rates in NaturalWaters[M].3rd ed.New York: John Wiley&Sons,Inc.,1996.
[12]Yoder C H,Pasteris JD,Krol K A,et al.Synthesis,structure,and solubility of carbonated barium chlor-and hydroxylapatites[J].Polyhedron,2012,44(1):143-149.
[13]Nriagu JO.Lead orthophosphates—IV Formation and stability in the environment[J].Geochimica et Cosmochimica Ac-ta,1974,38(6):887-898.
[14]Sternlieb M P,Pasteris JD,Williams B R,et al.The structure and solubility of carbonated hydroxyl and chloro lead apatites[J].Polyhedron,2010,29(11):2364-2372.
[15]Narasaraju T SB,Rao K K,Rai U S.Determination of solubility products of hydroxylapatite,chlorapatite,and their solid solutions[J].Canadian Journal of Chemistry,1979,57 (15):1919-1922.
Dissolution and solubility of alforsite[Ba5(PO4)3Cl]in aqueous solution
LIN Ju1,ZHU Zong-qiang1,PAN Yan2,WEICai-chun1,3,ZHU Yi-nian1,HE Li-na1
(1.a.Cooperative Innovation Center forWater Pullution Control and Water Security in Karst Area;b.Guangxi Key Laboratary of Environmental Engireering,Protection and Assessment,Guilin University of Technology,Guilin 541004,China;2.Environmental Monitoring Center of Guangxi,Nanning 530028,China;3.College of Light Industry and Food Engineering,Guangxi University,Nanning 530004,China)
Alforsite[Ba5(PO4)3Cl]was synthesized and characterized.The dissolution of prepared solid in HNO3solution(pH=2.00),ultrapurewater(pH=5.60)and NaOH solution(pH=9.00)were experimentally studied at25℃.The results indicated that the change of the solution pH values and element concentrations with time was greatly related to the initial pH of Se solution.The dissolution processes should include protonation,release and complexation of Ba2+,POand Cl-,recrystallization,etc.The average solubility products (Ksp)for alforsite[Ba5(PO4)3Cl]were determined to be 10-42.85,10-43.44and 10-43.34for the dissolution at 25℃ with initial pH of2.00,5.60 and 9.00,respectively.The corresponding Gibbs free energies of formation[ΔG fo]were calculated to be-6 235.82 kJ/mol,-6 239.16 kJ/mol and-6 238.58 kJ/mol.Stability of some common chlorapatites exists in the order of Pb5(PO4)3Cl>Cd5(PO4)3Cl>Ca5(PO4)3Cl>Ba5(PO4)3Cl>Sr5(PO4)3Cl>Zn5(PO4)3Cl.
alforsite;dissolution mechanism;solubility product;Gibbs free energy of formation
X53;O645.12
:A
2015-09-16
国家自然科学基金项目 (41263009;51638006);广西自然科学基金项目 (2014GXNSFBA118054);广西科学研究与技术开发计划项目 (桂科攻14124004-3-3);广西高校科学技术研究重点项目 (KY2015ZD053);广西矿冶与环境科学实验中心项目 (KH2012ZD004);广西高等学校高水平创新团队及卓越学者计划项目 (002401013001)
林 局 (1990—),男,博士研究生,环境科学与工程专业,linjucd@163.com。
朱宗强,博士,zhuzongqiang@glut.edu.cn。
林局,朱宗强,潘艳,等.磷氯钡石[Ba5(PO4)3Cl]在水溶液中的溶解作用及溶度积[J].桂林理工大学学报,2016,36(4):792-798.
1674-9057(2016)04-0792-07
10.3969/j.issn.1674-9057.2016.04.024
