FBN1新生突变引起的马凡综合征及其基因型-表型的关联研究*
2016-04-15潘啟豪梁小碧朱浚文王一鸣
潘啟豪, 梁小碧, 郭 勇, 朱浚文, 胡 彬, 王一鸣
(中山大学 1中山医学院医学遗传学教研室, 2疾病基因组研究所,广东 广州 510080; 3广州市妇女儿童医疗中心,心脏中心,广东 广州 510120; 4广州市公安局白云区分局刑警大队技术中队,广东 广州 510420; 5广州市公安局黄埔区分局,广东 广州 510700; 6中山大学新华学院,广东 广州 510520; 7华大基因,广东 深圳 518083)
FBN1新生突变引起的马凡综合征及其基因型-表型的关联研究*
潘啟豪1, 2,梁小碧3,郭勇4,朱浚文5,胡彬1, 2,王一鸣6, 7△
(中山大学1中山医学院医学遗传学教研室,2疾病基因组研究所,广东 广州 510080;3广州市妇女儿童医疗中心,心脏中心,广东 广州 510120;4广州市公安局白云区分局刑警大队技术中队,广东 广州 510420;5广州市公安局黄埔区分局,广东 广州 510700;6中山大学新华学院,广东 广州 510520;7华大基因,广东 深圳 518083)
[摘要]目的: 本研究对2个不同马凡综合征(Marfan syndrome)的小家系进行致病基因FBN1的编码区和剪切位点突变检测,以寻找致病的突变,并初步探索马凡综合征基因型-表型的关联。方法: 通过临床检查、实验室检查及心脏超声检查确诊2个无血缘关系的家庭中原疑似为马凡综合征的3例患者。运用新一代测序对家系1的疑似患者行FBN1基因的全外显子组测序,并对检出的致病性遗传变异进行Sanger验证及在所有家系成员中验证;对于家系2的存活成员,本研究直接进行PCR扩增FBN1基因的所有编码区及剪切位点,对产物进行直接Sanger测序。另外在50个正常对照中对新发现的突变位点进行基于PCR产物的测序分析,以排除多态性;并对实验结果行生物信息学分析。结果: 所有存活的疑似患者均确诊为马凡综合征。在家系1中,我们检测到了一个FBN1基因数据库中尚未报道的新突变c.4685G>A(p.Cys1562Tyr),并且患者父母和同胞姐姐均未检测到此变异,故此突变为一个新生突变。该错义突变使第1 562位上极性中性的含硫的半胱氨酸被极性中性的含羟苯基的酪氨酸所替代,影响了fibrillin-1蛋白一个TGF-β结合结构域,导致蛋白质的二级结构发生改变。家系2含父母及一对同卵双胎患者,其中一患者已去世。我们在存活患者检测到1个FBN1基因的已报道致病突变c.3706T>C (p.Cys1236Arg),该突变在患者父母中不存在,故也为新生突变。结论: 本文报道了一例FBN1基因的新突变及另一例由FBN1基因已知突变引起的马凡综合征,二者皆为新生突变,并在家系中进行了基因型-表型的比较,表明家系1的新突变可能与经典马凡综合征的表型相关,而家系2的已知突变确和新生儿重症马凡综合征表型相关。
[关键词]马凡综合征; FBN1基因; 新生突变; 基因型; 表型
马凡综合征(Marfan syndrome)是一种常染色体显性遗传的全身性结缔组织病,其发病率约为1/5 000[1],临床表现主要有蜘蛛指/趾、脊柱侧弯,主动脉病变,晶状体异位及硬脑膜膨出等。在遗传学上,绝大部分的马凡综合征致病基因定位于15q21.1的FBN1,该基因包含有65个编码外显子,编码原纤维蛋白1(fibrillin-1)。Fibrillin-1有47个上皮生长因子样结构域和9个转化生长因子β结合结构域(transforming growth factor-β binding domain, TB domain),是细胞外基质结构组成部分,主要参与细胞生长与维持。它主要表达部位在血管、软骨、肾脏、肺部、皮肤、尿道以及人的骨骼中和骨细胞,是眼球晶状体悬韧带的主要成分。FBN1基因的错义突变和显性负效应是导致细胞外基质fibrillin-1缺陷的主要遗传因素。
本研究收集了2个马凡综合征的家系进行研究。对于家系1,我们对先证者先行二代测序再行Sanger验证;家系2含2例患者为同卵双胎,其中1例已病故,我们对存活患者进行FBN1基因所有编码区及剪切位点的突变检测。检测结果均在家系中其他成员进行验证,现将结果报道如下。
材料和方法
1研究对象
本研究纳入的2个无血缘关系的小家系(图1)均来自广州市妇女儿童医疗中心,同时纳入了50例无血缘关系的正常人作为对照组。所有家系成员均经过详细的病史采集及体格检查,所有患者均行心脏彩超检查。马凡综合征的诊断符合Ghent诊断标准。家系1 中的先证者和家系2 中的先证者及其同卵双生的弟弟被诊断为患者,其余家庭成员均为无发现马凡综合征的正常人。本研究符合“2013年修订的赫尔辛基宣言原则”并取得了中山大学中山医学院伦理委员会的批准,所有的研究对象均签署了知情同意书(小于18周岁的研究对象由其监护人签署知情同意书)。
家系1患儿13岁,因“发现心脏异常1周”就诊,活动耐量与正常小孩无异,当地医院查有“晶状体脱位”,其余体查无明显异常,行心脏彩超提示“主动脉窦部明显扩张(最宽处内径36 mm)并局限返流”。
家系2患儿为7月大的足月儿,因“气促10 d,咳嗽2 d”住院治疗,体查可见其身长较长,胸廓呈鸡胸,胸骨隆起,双肺呼吸音增粗,偶可及干湿啰音。心律齐,心音稍钝,心率142 min-1,心尖区可及收缩期2/6级柔和杂音,无传导。双上肢肘关节内翻,活动度正常,手指、脚趾细长。行胸片提示“脊柱侧弯”,行心脏彩超提示 “主动脉窦部扩张(最宽处内径22 mm)”,见图2。其一双胞胎哥哥于3月大时被诊断为“马凡综合征、呼吸衰竭”,4月大时死于多器官功能衰竭。随访该患儿,其出院后因反复感染入住不同医院,并于1岁5月死于多器官功能衰竭。
2方法
2.1基因组DNA的提取取所有成员外周血200 μL,根据Tiangen血液基因组小量抽提试剂盒方法抽提DNA。
2.2引物设计及PCR扩增利用UCSC数据库(http://genome.ucsc.edu)获取FBN1基因的序列信息。利用Oligo 6.0软件设计 65对引物,覆盖所有编码区域及剪切位点。所有引物均由深圳华大基因研究院(BGI tech)合成,需要引物序列的读者可向作者索取。PCR扩增反应体系为普通30 μL体系:10×Taq buffer 3 μL,25 mmol/L MgCl22.5 μL,2.5 mmol/L dNTP mixture 3 μL,3.2 μmol/L上、下游引物各1 μL,Taq DNA polymerase 1 μL(Thermo),DNA模板12.3Sanger测序及突变分析鉴定应用ABI 3730XL DNA测序仪对所有PCR产物行直接测序;应用软件Sequence Scanner 1.0(Applied Biosystems)对测序结果进行分析判读[2]。突变命名参考标准的序列变异命名法则(http://www.hgvs.org/mutnomen);将检测到的遗传突变与人类基因组突变数据库(http://www.hgmd.cf.ac.uk)进行比对并查PubMed上已发表的文章确定所检突变是否为已知突变。对于错义突变的位点,利用Clustal Omega program(http://www.uniprot.org/align/)进行物种保守性比对,并利用SIFT软件进行功能学预测。
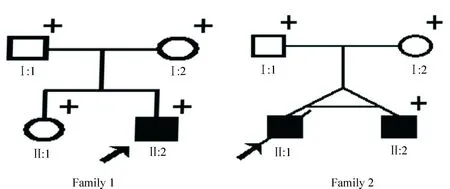
Figure 1.Pedigree of 2 families with Marfan syndrome was shown. A filled square indicated an affected male. The proband is marked by an arrow.
图12个马凡综合征家系的家系图
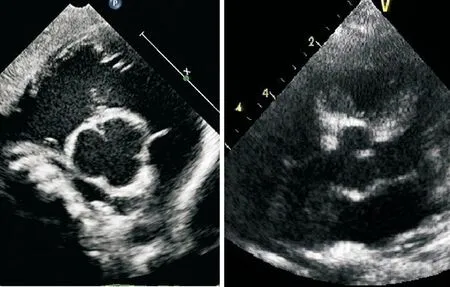
Figure 2.Echocardiogram of the patient (II:2) in the family 2.
图2家系2患者(II:2)的心脏超声影像
结果
1家系1突变检测结果
本家系患者二代测序[3]及直接测序结果发现一个位于第37外显子且预测为“DAMAGING”的错义突变c.4685G>A(p.Cys1562Tyr),见图3,该突变未见报道且在家系其余成员中均未检出该突变,为新的denovo突变。它使得第1 562位上极性中性的含硫的半胱氨酸被极性中性的含羟苯基的酪氨酸所替代,影响了fibrillin-1蛋白的1个TB domain,导致蛋白质的二级结构发生改变。
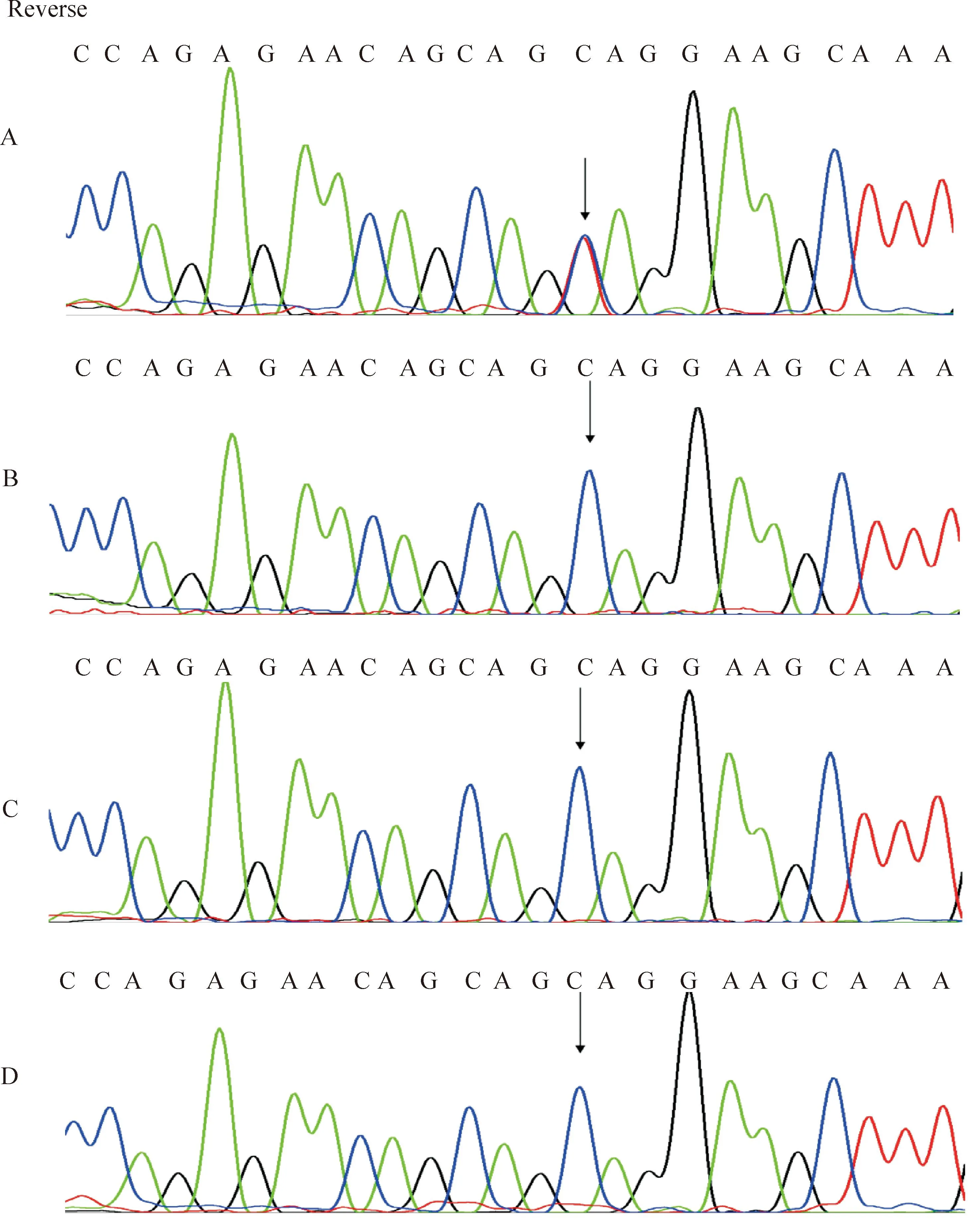
Figure 3.Partial sequencing results of patient II:2 (A), his parents (B, C) and his sister (D) in family 1. The arrows indicate the position of the mutation c.4685G>A (p.Cys1562Tyr).
图3家系1各成员的c.4685G>A (p.Cys1562Tyr)突变检测结果
2家系2突变检测结果
本家系存活患者的直接测序结果发现了一个位于第29外显子且预测为“DAMAGING”的错义突变c.3706T>C (p.Cys1236Arg),见图4,是已报道的致病突变并被收录在HGMD数据库中。该突变在表型正常的父母中均未检出该突变,为denovo突变。它使得第1 236位上极性中性的含硫的半胱氨酸被碱性氨基酸的精氨酸所替代,影响了fibrillin-1蛋白的其中一个EGF-like calcium-binding 19 domain,导致了蛋白质功能的缺陷。
3突变氨基酸在物种间的保守性比对结果
2个家系中发现的2个错义突变c.4685G>A(p.Cys1562Tyr)和c.3706T>C(p.Cys1236Arg)在氨基酸水平进行人类、猩猩、牛、大鼠以及小鼠这6种生物的物种保守性比对的结果显示,被替代的氨基酸均发生在高度保守的氨基酸区域,见图5。
讨论
本研究对2个马凡综合征的小家系进行了FBN1基因的突变检测,发现患者均携带FBN1基因的致病性突变。家系1的先证者中发现的错义突变c.4685G>A(p.Cys1562Tyr)为一个国际上未报道的新的新生突变。Biggin等[4]在对57个无关个体的马凡综合征患者进行FBN1基因的测序结果中发现,一个表现为下肢长度比例增加、关节活动度增大、上腭狭窄特征的面部表型以及主动脉扩张的患者携带的错义突变c.4691G>A(p.Cys1564Tyr)在我们家系1中发现的新突变附近,且影响了TB结构域,因此佐证了我们对家系1新突变致病性的推断。而在家系2的患者中发现的错义突变c.3706T>C(p.C1236R)为一个已报道的新生突变,该突变被认为与新生儿马凡综合征发病相关。

Figure 4.Partial sequencing results of patient II:2 (A) and his parent (B, C) in family 2. The arrows indicate the position of the mutation c.3706T>C (p.Cys1236Arg).
图4家系2各存活成员的c.3706T>C (p.Cys1236Arg) 突变检测结果
结合两患儿的发病年龄、病情进展及预后,家系1的患儿发病较晚、病情进展缓慢、预后较好,符合经典的马凡综合征;家系2的患儿及其同胞哥哥发病早、病情进展迅速、死亡率高,符合新生儿马凡综合征的诊断。经典的马凡综合征患者通常青春期或者成年后起病,如治疗及时,预后相对较好;新生儿马凡综合征是马凡综合征中极其罕见的一种分型,这类患儿病情进展迅速,累及全身多器官,约一半的患儿在一岁左右死于心脏衰竭或其它系统衰竭[5]。Kirschner等[6]研究指出,经典马凡综合征与新生儿型马凡综合征表现出fibrillin-1蛋白酶易脆性以及蛋白功能的差异。与经典马凡综合征或野生型fibrillin-1相比,具有新生儿马凡综合征突变的fibrillin-1更易于被溶蛋白酶裂解且影响了与肝素/硫酸乙酰肝素的相互作用,阻碍了微纤维的正确装配。Stheneur等[7]的研究发现,发生在FBN1第24~32外显子的突变通常与新生儿马凡综合征相关。他们还认为位于半胱氨酸区域的突变携带者发生晶状体脱位的比例显著高于不位于半胱氨酸区域携带者[8]。在本研究的两个家系中,家系1患儿查体有“晶体脱位”,家系2患儿因其突出的呼吸循环障碍的表型,并未查眼部情况。
马凡综合征的临床表型具有多变性[9],且其基因型-表型的关系并未被充分的研究。由于FBN1突变引起的马凡综合征会导致全身性结缔组织病,影响患者的日常活动甚至生活质量;且该疾病具有家族性传递的可能;此外新生儿马凡综合征患儿往往于婴幼儿期死于严重的呼吸循环衰竭,对患儿家属构成极大地精神打击。因此,无论对于家族性的马凡综合征以及不典型的或者尚未发病的携带者,或是对于患有主动脉扩张等心血管畸形的婴幼儿,我们建议行马凡综合征的遗传学筛查[11]。若遗传结果为阳性,应与患者及患者家属在疾病的预后及疾病的进程方面进行有效地沟通,促进医患的配合并尽最大努力控制疾病的发展,预防严重并发症的发生。

Figure 5.The sequence alignment of FBN1 protein among different species. The results of human, chimpanzee, bovine, rat and mouse were showed. The red frame in the alignment showed the amino acid affected by the mutation.
图5FBN1突变氨基酸序列的物种保守性比对的结果
由于denovo突变主要有两种来源可能,即胚胎发育过程中产生的,或为父母生殖细胞嵌合体。生殖细胞嵌合体是指当突变只发生在胚胎早期生殖细胞形成过程中,这种携带突变的细胞系只占生殖细胞腺的一部分,机体其它组织细胞不携带突变,因此在对外周血提取的基因组中可能不携带该突变或该突变携带的细胞比例太少不被检出[10]。对于这类患者家庭,我们建议行相关产前遗传学诊断。
最后,本研究比较了2个不同的马凡综合征家系患者的FBN1基因突变筛查的结果,扩大了FBN1基因的突变谱。初步分析了基因型与表型特点、疾病进程及预后的联系,为查有新生突变病史的家庭,特别是携带新生儿马凡综合征突变的患儿家庭,进行早期的确诊和干预,并提供遗传咨询以及为其后代的临床及基因检测提供依据。
[参考文献]
[1]Yang RQ, Jabbari J, Cheng XS, et al. New population-based exome data question the pathogenicity of some genetic variants previously associated with Marfan syndrome[J]. BMC Genet, 2014, 15:74.
[2]钟良英,丁红珂,袁萍,等.EXT2基因突变引起的多发性骨软骨瘤的遗传学及表型分析[J]. 中国病理生理杂志, 2011, 27(5):980-984.
[3]Xiao Y, Liu Y, Yang K, et al. Next generation sequencing as a rapid molecular diagnosis for Marfan syndrome in a Chinese family with mutations in thefibrillin-1 gene[J]. Clin Chim Acta, 2015, 439:58-60.
[4]Biggin A, Holman K, Brett M, et al. Detection of thirty novelFBN1 mutations in patients with Marfan syndrome or a related fibrillinopathy[J]. Hum Mutat, 2004, 23(1):99.
[5]Kochilas L, Gundogan F, Atalay M, et al. A novel mutation of the fibrillin-1 gene in a newborn with severe Marfan syndrome[J]. J Perinatol, 2008, 28(4):303-305.
[6]Kirschner R, Hubmacher D, Iyengar G, et al. Classical and neonatal Marfan syndrome mutations in fibrillin-1 cause differential protease susceptibilities and protein function[J]. J Biol Chem, 2011, 286(37):32810-32823.
[7]Stheneur C, Faivre L, Collod-Beroud G, et al. Prognosis factors in probands with anFBN1 mutation diagnosed before the age of 1 year[J]. Pediatr Res, 2011, 69(3):265-270.
[8]Liang C, Fan W, Wu S, et al. Identification of a novel FBN1 mutation in a Chinese family with isolated ectopia lentis[J]. Mol Vis, 2011, 17:3481-3485.
[9]Arbustini E, Grasso M, Ansaldi S, et al. Identification of sixty-two novel and twelve known FBN1 mutations in eigh-ty-one unrelated probands with Marfan syndrome and other fibrillinopathies[J]. Hum Mutat, 2005, 26(5):494.
[11]Radke RM, Baumgartner H. Diagnosis and treatment of Marfan syndrome: an update[J]. Heart, 2014, 100(17):1382-1391.
(责任编辑: 林白霜, 罗森)
Twodenovomutations including 1 novel mutation in FBN1 and genotype-phenotype correlation in 2 Chinese Marfan syndrome families
PAN Qi-hao1, 2, LIANG Xiao-bi3, GUO Yong4, ZHU Jun-wen5, HU Bin1, 2, WANG Yi-ming6, 7
(1DepartmentofMedicalGenetics,ZhongshanSchoolofMedicine,2CenterforGenomeResearch,SunYat-senUniversity,Guangzhou510080,China;3HeartCenter,GuangzhouWomenandChildren’sMedicalCenter,Guangzhou510120,China;4DepartmentofCriminalInvestigationBrigadeofBaiyunBranch,GuangzhouPublicMunicipalPublicSecurityBureau,Guangzhou510420,China;5HuangpuBranchofGuangzhouPublicMunicipalPublicSecurityBureau,Guangzhou510700,China;6XinhuaCollege,SunYat-senUniversity,Guangzhou510520,China;7BeijingGenomicsInstituteinShenzhen,Shenzhen518083,China.E-mail:ywzhong@hotmail.com)
[ABSTRACT]AIM: To investigate the genetic cause of 2 Chinese families with Marfan syndrome. METHODS: The clinical and laboratory investigations were performed in the 2 unrelated Chinese families. Family 1 had 1 patient with cardiac problem. Family 2 had 2 patients: one died, and the other with respiratory and cardiac problems. Next generation sequencing and Sanger sequencing in the Marfan syndrome causal gene FBN1 were performed in the patient, his unaffected sister and the parents of family 1. Sanger sequencing covering all the exons and intron-exon boundaries were performed in the patient and the parents in family 2. Bioinformatic analysis was engaged in the variations unravelled. Fifty healthy individuals were also investigated in the same manner. RESULTS: Both patients were diagnosed with Marfan syndrome. A novel mutation c.4685G>A (p.Cys1562Tyr) was detected in the patient of family 1 but was absent in his parents and the unaffected sister. This is a previously unreported novel mutation. In the mutation a conserved Cys was substituted by a Tyr in amino acid 1562 affecting a TGF-β binding domain and the secondary structure in the encoded protein. We also detected the mutation c.3706T>C (p.Cys1236Arg) in the patient of family 2. It was absent in the unaffected parents, and therefore was a de novo mutation too. This mutation has been previously reported and known to be associated with neonatal Marfan syndrome. Both mutations were absent in the 50 healthy controls. We also compared the genotype and phenotypes of the 2 families. CONCLUSION: We report 2 de novo mutations in 2 Chinese families with Marfan syndrome. One of the 2 mutations is novel. The phenotype of the mutation c.4685G>A(p.Cys1562Tyr) in family 1 is associated with classical Marfan syndrome, while that of c.3706T>C (p.Cys1236Arg) in family 2 is with neonatal type of Marfan syndrome. De novo mutations may be a cause for a proportion of mutations underlying the disease. The novel mutation also expends the mutational spectrum of the FBN1 gene.
[KEY WORDS]Marfan syndrome; FBN1 gene; De novo mutation; Genotype; Phenotype
doi:10.3969/j.issn.1000- 4718.2016.03.023
[中图分类号]R363
[文献标志码]A
通讯作者△Tel: 020-87332055; E-mail: ywzhong@hotmail.com
*[基金项目]国家自然科学基金资助项目(No. 31471193)
[收稿日期]2015- 09- 14[修回日期] 2015- 12- 14
[文章编号]1000- 4718(2016)03- 0527- 07
杂志网址: http://www.cjpp.net
