褐煤模型化合物中官能团持水性质的理论研究
2016-04-11张向飞孙铭骏郭娟娟曹泽星厦门大学化学化工学院福建厦门36005中国矿业大学化工学院江苏徐州6
张向飞,孙铭骏,郭娟娟,冯 莉,曹泽星*(.厦门大学化学化工学院,福建厦门36005;.中国矿业大学化工学院,江苏徐州6)
褐煤模型化合物中官能团持水性质的理论研究
张向飞1,孙铭骏1,郭娟娟1,冯 莉2,曹泽星1*
(1.厦门大学化学化工学院,福建厦门361005;2.中国矿业大学化工学院,江苏徐州221116)
摘要:采用密度泛函方法,研究了褐煤分子中官能团与水分子的相互作用;确定了不同官能团持水性的相对强弱,讨论了水分高低和结构环境对其持水性和脱水过程的影响,建立了脱水过程近似热力学关系式.计算结果表明,单个官能团的持水性呈现一定的饱和度,其周围化学环境对持水能力影响显著.压力增加会提高脱水温度,但影响不显著,加压有助于水分以水团簇形式脱去,降低脱水能耗.
关键词:密度泛函计算;褐煤;持水官能团;脱水;热力学公式
我国煤炭资源丰富,储量占世界石化能源的一半以上.然而,高含灰、高含硫、高含水的低品质煤约占煤炭资源总量的40%,其中低品质褐煤约占总储量的14%[1].褐煤具有挥发分高、含氧量高、稳定性差、含氮硫等易形成污染物的元素相对较少的特点,其中含有的原生腐植酸是其区别于其他煤种的主要特征组分[2].由于褐煤中水分的质量分数较高,可达30%~ 60%[3],若直接燃烧利用,会产生各种不利因素:水分蒸发带走大量热能;易与含硫、羧基等酸性官能团的挥发成分形成具有腐蚀性的冷凝液等[4].同时,干燥后的褐煤易发生复吸现象,影响干燥效果.因此,实现低耗能高效脱水并有效抑制复吸的发生对褐煤提质与优化利用具有十分重要的意义.
褐煤中赋存的水通常具有3种形式:游离水或表面微弱结合的水、在微孔中或微粒间隙中强吸附的水和以化学吸附形式存在的水[5].前2种水分通过抽气、适当升温和加压可以脱去,第3种水分同褐煤中含有的各类官能团间存在较强的相互作用,脱水过程需要在较高的温度下才能进行.褐煤较发达的孔隙结构和丰富的含氧官能团是导致其含水量高和易复吸的主要因素;此外还存在着少量含氮、硫的官能团,这些官能团极性较强,极易形成氢键把水分子结合在煤表面[6].长期以来,褐煤干燥的提质实验研究在国内受到广泛重视[7-12],然而,由于煤组成与结构的复杂性,在原子/分子上的理论研究相对较少[3,13].本文试图通过对煤结构单元模型与水分子相互作用的理论计算,预测褐煤分子中官能团的持水性质,为探明不同脱水过程的热力学特征提供理论支持.
1 计算方法
由于褐煤分子中官能团与水分子间是一种弱相互作用,选取常用的B3LYP、M06、ωB97XD、M06-2X 4种泛函,在6-311+G(d,p)基组水平下进行了测试计算.表1给出了不同泛函预测的1-苯基-2-丙酮与1个水分子形成氢键复合体系的结合能(ΔE)和结构.先前的研究表明[14-16],M06-2X杂化泛函对非共价相互作用及中程电子相关效应有较好的描述.如表1所示,这里M06-2X计算优化的构型不仅呈现出水与CO的氢键作用,还包括了水的—OH与苯环π电子间的弱相互作用,其预测的ΔE最大.
在常温常压(298.15 K,101.3 k Pa)的气相条件下,采用M06-2X/6-311+G(d,p)方法,优化了褐煤模型化合物中含氧、氮、硫等官能团与1~4个水分子相互作用形成复合物的可能平衡构型,结合频率计算,讨论了结构的稳定性并对预测的能量进行了零点能校正.由于褐煤的成分及其结构十分复杂,难以构造出统一的结构模型来表示褐煤分子[17].考虑到计算上的可行性,我们截取了一些具有代表性的褐煤分子片段作为研究对象,主要考察各种官能团持水性质及脱附能的大小,并借助热力学循环,讨论了压力对脱水性质的影响.所有计算采用Gaussian 09 B.01程序[18].
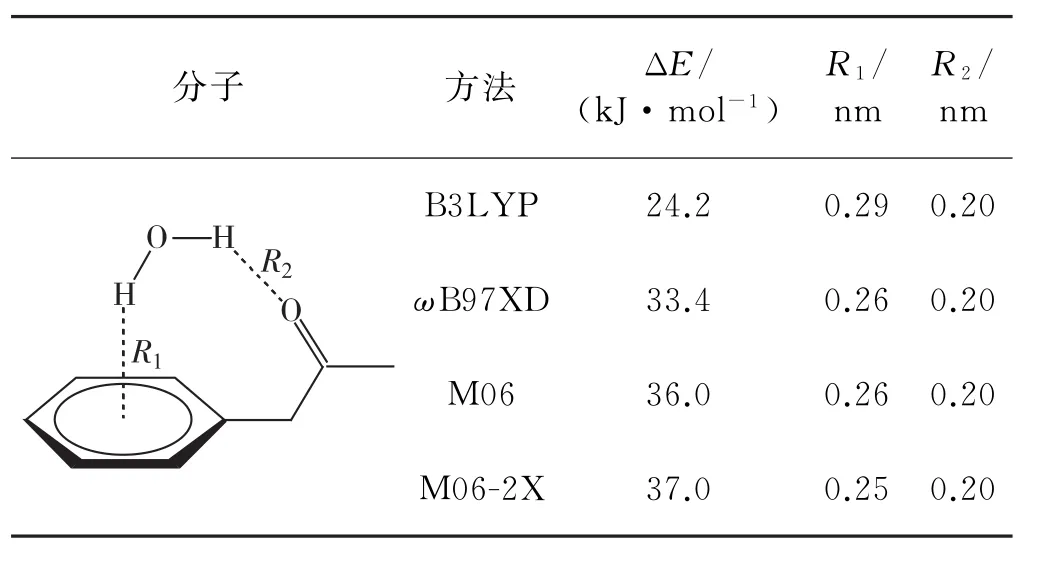
表1 不同方法预测的氢键键长(R1和R2)与ΔETab.1 Predicted hydrogen-bond lengths (R1,R2)andΔE by different approaches
2 结果与讨论
2.1官能团的持水性质
褐煤分子中存在着许多不同官能团,我们选取具有代表性的官能团—COOH、—OH、—CO、—O—、—N—、—S—等,并研究这些官能团在不同化学环境下与1~4个水分子的相互作用.水分子通过氢键相互作用环绕在官能团周围,脱水过程表示为:
Lignite…n H2O→Lignite+(H2O)n/n H2O.
其中:(H2O)n为水团簇,即水分子之间形成了氢键; n H2O表示n个单独脱去的水分子.计算结果见表2.从表中可以看出:当水分以单个气态水分子脱去时(即脱去的多个水分子之间无氢键相互作用),需要更多的能量;而脱去水分为水簇形式时,需要的能量相对少得多.这一脱水能耗的差别归结为水团簇中的水分子间氢键相互作用可以补偿破坏官能团与水分子之间的相互作用.
这些官能团脱去1个水分子所需要的能量一般在20.9~33.5 kJ/mol之间,与水分子之间形成的氢键强度相当,因此,将导致官能团的持水与吸附水分子的成簇作用形成竞争,从而使不同官能团的持水性与水分的多少及官能团的协同作用密切相关.我们注意到,—COOH(羧基)官能团与水分子的结合能高达50.2 kJ/mol,这是因为1个羧基与1个水分子之间能形成2个氢键.表2结果显示,这些官能团脱附2个团簇水时需要较多的能量,而脱去4个(或3个)团簇水时,大部分能量反而降低.这些结果反映单个官能团吸附2(或3个)水分子时,已经达到饱和,更高的水分倾向形成水团簇;同时也说明,在褐煤脱水过程中,随着未饱和持水官能团的增加,极易对水分产生复吸,造成褐煤脱水提质的困难.

表2 常温常压下脱水过程的热力学焓变(ΔH)和自由能变化(ΔG)Tab.2 Predicted thermodynamic values of the dewatering process under the room temperatures and pressures
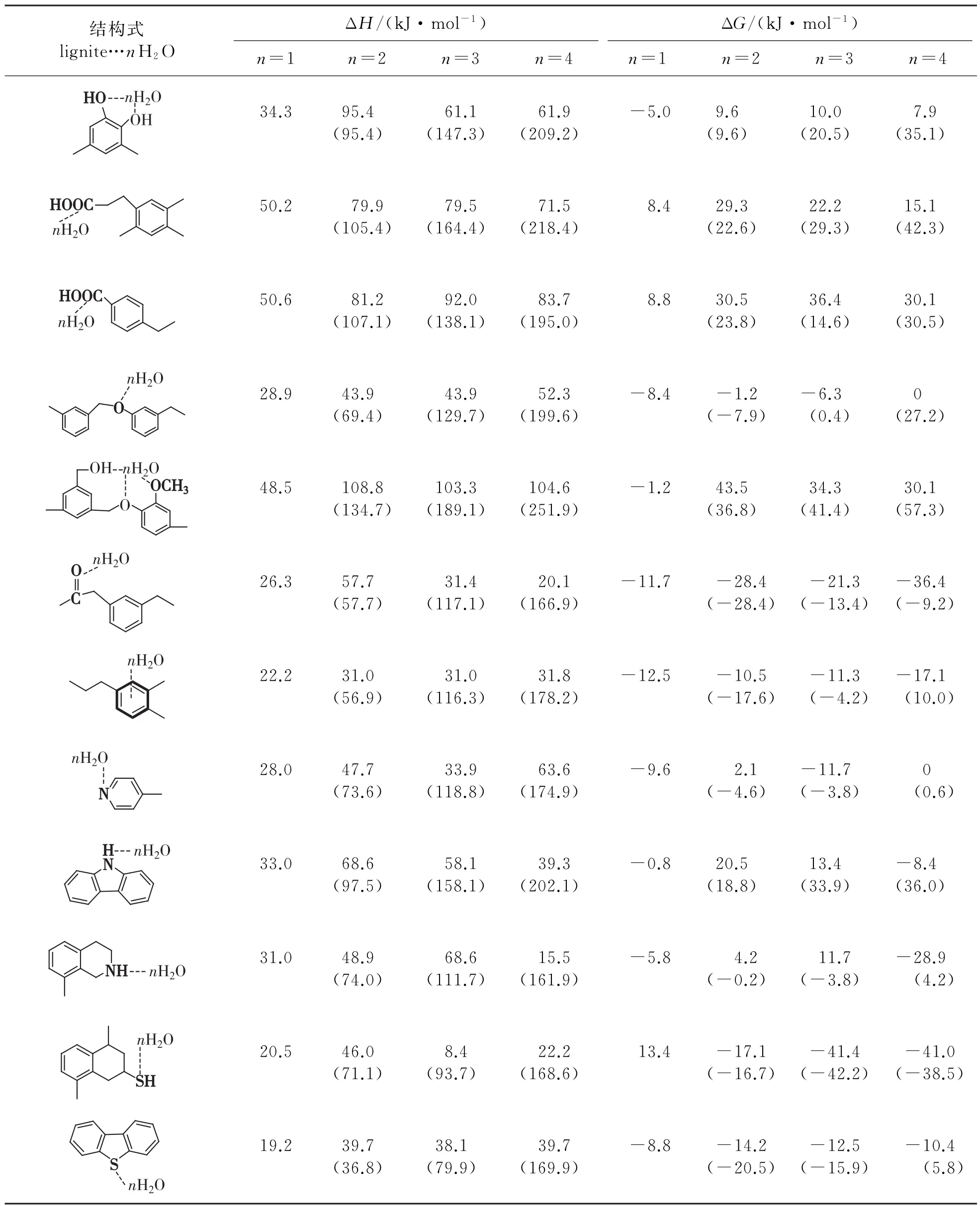
续表
褐煤中存在着大量芳香性的苯环结构单元,但很少有萘、蒽、菲类联苯结构,因此我们以苯分子结构模型,讨论水与芳香π电子的相互作用.当水分子接近时,苯环大π键π66电子与水分子中的氢存在非共价相互作用,表现为静电吸引.如图1所示,水分子中的2个氢原子距苯环中心0.26 nm,ΔE为16.3 kJ/mol.当苯环同侧有2个水分子存在时,会远离苯环而优化成1个水团簇,从而表现出一定的疏水性,脱去这个水团簇仅需能量5.0 kJ/mol.当2个水分子分别在苯环的两侧时,距苯环中心的距离都约为0.26~0.29 nm,脱去2个水分子消耗能量为30.1 kJ/mol,近似为脱去1个水分子的2倍.计算表明:苯环两侧都能与水分子H端相互作用,但第2个水分子的加入未能加强苯环的持水性.

图1 苯环吸附水分子(n H2O)的平衡构型(单位:nm)Fig.1 Optimized structures of molecular complexes of benzene with water molecules(n H2O)(unit:nm)

图2 咔唑与水分子(n H2O)形成复合物的平衡构型(单位:nm)Fig.2 Optimized structures of molecular complexes of carbazole with water molecules(n H2O)(unit:nm)
当苯环上存在供电子基团时,会加强其与水的相互作用.如图2所示,咔唑中的氮原子由于与两边苯环形成了共轭大π键,其持水性很弱,但加强了咔唑共轭大π键对水分子的作用,共轭环与单个水分子相互作用的ΔE为25.5 kJ/mol,和—NH—的持水能力接近(表2:31.0~33.0 kJ/mol).因此,尽管疏水性的苯环与水分子的相互作用相对较弱,但褐煤中大量这类共轭环的存在及其邻近供电子基团的影响,也会对脱水过程中的能耗产生一定的影响.
褐煤中的硫主要以矿物硫和有机硫的形式存在,其中有机硫主要以二苯并噻吩的形式存在.这里我们以二苯并噻吩、苄硫醇为结构模型,研究了含硫官能团与水的相互作用,图3给出了含硫模型体系与水分子形成分子复合物的可能结构.
对于二苯并噻吩,硫原子参与邻近苯环共轭成键作用,使得水分子不能与硫原子形成直接有效的氢键相互作用,而与苯环大π键形成非共价相互作用,其ΔE为19.2 kJ/mol.若在二苯并噻吩同侧加入第2个水分子,2个水分子易形成团簇;若在异侧,水分子距苯环中心的距离为0.27 nm,脱去这2个水分子所需的ΔE为39.7 kJ/mol.对于苄硫醇,水分子与巯基形成的氢键键长达到了0.25 nm,远比其他官能团形成的氢键长.图3显示此时还包含芳环氢与水中氧的相互作用,脱去这个水分子所需的ΔE为19.2 kJ/mol.计算显示,2个水分子与苄硫醇的ΔE为44.3 kJ/mol,说明第2个水分子会加强巯基与水的相互作用,和其他官能团持水性不同.
表2的结果还表明:同种元素形成的不同官能团,其持水性质也不尽相同,比如含氮的官能团,吡啶持水能力最强;而且同一种官能团在不同环境下,例如醇羟基与酚羟基,对水的吸附能也有差别.可见,官能团周围化学环境对水的吸附作用影响很大.总体上,在没有其他官能团的协同作用下,单个官能团吸附水的能力强弱顺序为:羧基>吡啶>羟基>氨基>酮基>苯环>巯基.对于大多数官能团来说,当其附近还有其他官能团时,其持水性质会发生变化,例如在醇羟基旁边有羧基存在时,羟基对水的吸附大大增强(如图4),吸附能增加了近41.8 kJ/mol,这是因为羧基也直接参与了和水分子间的氢键相互作用.同样,单个醚氧官能团脱水需要的能量是25.0~50.0 kJ/mol,但是当其附近还有其他持水官能团存在时,脱水需要的能量增加了1倍.

图3 二苯并噻吩和苄硫醇与水分子(n H2O)形成复合物的平衡结构(单位:nm)Fig.3 Optimized structures of dibenzothiophene and benzyl mercaptan binding water molecules(n H2O)(unit:nm)
2.2 温度和压力对官能团持水性质的影响
褐煤分子片段的脱水过程可近似地表示为

其中A为结合了n个水分子的褐煤分子片段,B为脱附水分子后的褐煤分子片段,s为该物质的状态为固态,g为气态.为了评估外界条件温度与压力对褐煤脱水性质的影响,可以构造如下热力学循环:
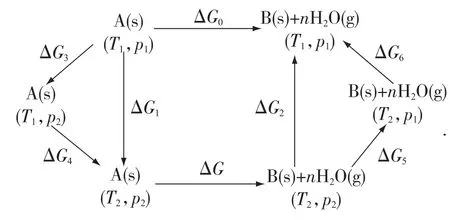
其中:ΔG0=ΔG1+ΔG+ΔG2,
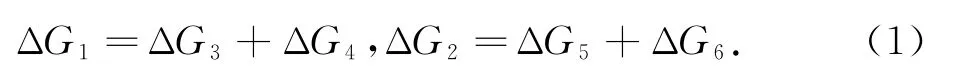
对于循环中的等温变压过程(其中气体均考虑为理想气体),有

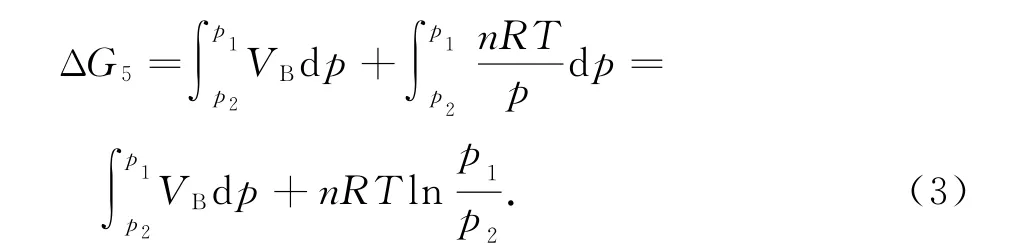
对于循环中的等压变温过程,有
ΔG=ΔH-Δ(TS)=ΔH-TΔS-SΔT,假定该过程近似为绝热可逆过程,则
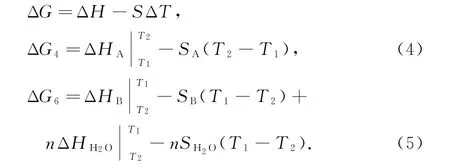
为了获得ΔG随着温度T与压力p变化的近似公式,假定褐煤脱水前后的分子A和B都是固体,无化学反应发生.取近似:

则有
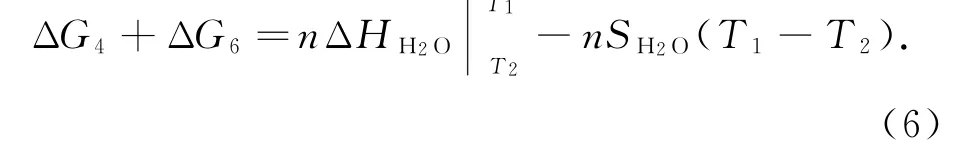
故面


图4 含羟基模型化合物与水分子(n H2O)形成复合物的平衡结构(单位:nm)Fig.4 Optimized structures of hydroxyl compounds binding water molecules(n H2O)(unit:nm)
若VA≈VB,上式可化为
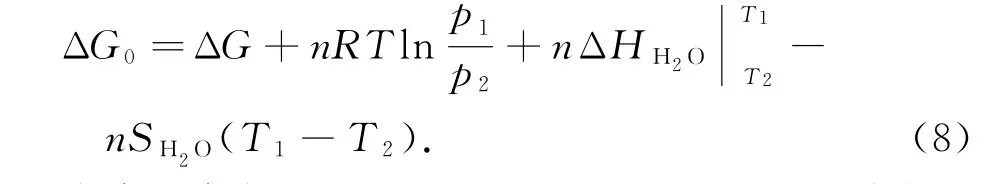
令参考条件:p1=101.3 k Pa,T1=298 K,脱水过程对应的自由能变化为ΔG0,则由热力学循环有任意温度与压力条件下的自由能变化为:
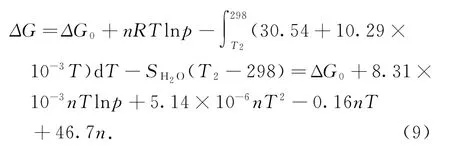
其中ΔG0为常温和常压下的吉布斯自由能变化,可以通过理论计算得到,例如由表2可知,羧基脱水的反应ΔG0=8.4 kJ/mol.脱水过程能自发发生的热力学条件是ΔG(T,p)≤0,由公式(9)可以预测不同压力下的临界脱水温度,相关结果列于表3.
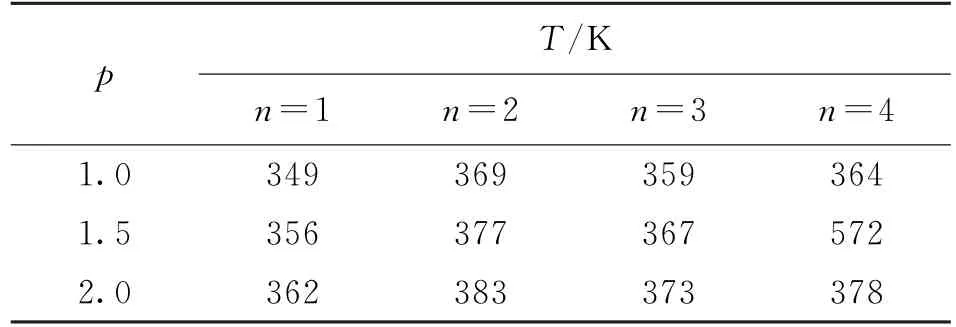
表3 不同压力下羧基脱水的临界温度Tab.3 The critical temperatures for the carboxyl dewatering under different pressures
由表3可知,随着压力的增加,脱水温度会逐渐升高,在相同的压力条件下,持2个水分子的羧基官能团(表2中第6行)脱水温度最高,这一现象可归结为持水相互作用焓变与脱水熵增效应的平衡.压力与脱水温度的关系可以在一定程度上反映褐煤局域结构环境,如孔道、不同界面等对脱水性质的影响.整体上,压力对脱水温度的升高并不显著,但有助于水分以水团簇的形式脱去,降低脱水的耗能(见表2).现有褐煤脱水干燥过程中,为了维持高温下水分不被汽化,普遍使用了过热蒸汽和加压技术[4].
3 结 论
通过密度泛函理论计算,我们较系统地讨论了褐煤分子片段模型中常见官能团的持水性质,确定了一些常见官能团与水分的相互作用能.计算表明单个官能团结合水的能力强弱依次为:羧基>吡啶>羟基>氨基>酮基>苯环>巯基.大多数官能团的持水能力受周围化学环境的影响较大,邻近官能团的协同作用可以显著提高其持水能力.对高水分体系,水以水团簇形式脱去,耗能低.基于热力学循环,我们建立了脱水过程自由能随温度、压力变化的热力学近似公式,计算结果显示脱水温度受压强的影响较小.本文获得的结果有助于在原子/分子水平上认识褐煤的持水性质,为进一步的理论和实验研究提供参考.
参考文献:
[1] 郭树才.煤化工工艺学[M].3版.北京:化学工业出版社, 2006:1-2.
[2] 陈鹏.中国煤炭性质、分类和利用[M].2版.北京:化学工业出版社,2007:17-24.
[3] ZHANG Z Q.Structure and dynamics in brown coal matrix during moisture removal process by molecular dynamics simulation[J].Mol Phys,2011,109(3):447-455.
[4] 蒋斌,高俊荣,贾世阳,等.褐煤干燥脱水技术的研究进展[J].干燥技术与设备,2011,9(2):64-68.
[5] ROZGONYI T G,赵振东.用饱和蒸汽加工提质高水分褐煤[J].煤炭转化,1987(2):64-71.
[6] ALLARDICE D J.The water in brown coal[M].Oxford: Burterworth-Heinemann,1991:102-150.
[7] 王宝俊,凌丽霞,赵清艳,等.气体与煤表面吸附作用的量子化学研究[J].化工学报,2009,60(4):995-1000.
[8] 傅爱萍,冯大诚,邓从豪.水在石墨(0001)面簇模型桥位上吸附的量子化学研究[J].高等学校化学学报,1998,19 (5):792-795.
[9] 李建武,白公正,雷宝林,等.吐哈盆地煤层的吸附性及其影响因素[J].煤田地质与勘探,2001,29(2):30-32.
[10] 王继仁,赵庆福,邓存宝,等.煤表面对多种气体分子混合吸附的微观机理[J].计算机与应用化学,2008,25(4): 390-394.
[11] KUMAGAI H,CHIBA T,NAKAMURA K,et al. Change in physical and chemical characteristics of brown coal along with a progress of moisture release[J].ACS Div Fuel Chem,1999,44(4):633-394.
[12] VU T,CHAFFEE A,YAROVSKY I.Investigation of Lignin-water interactions by molecular simulation[J]. Mol Simulat,2002,28:981-991.
[13] ZHANG Z T,TURNER C H.Water-induced interactions between boron-doped carbon nanotubes[J].J Phys Chem, 2014,118(31):17838-17846.
[14] 王新华,冯莉,曹泽星,等.取代基效应对褐煤模型化合物离解焓影响的理论研究[J].化学学报,2013,71(7): 1047-1052.
[15] BRYANTSEV V S,DIALLO M S,VAN DUIN A C,et al.Evaluation of B3LYP,X3LYP,and M06-class density functionals for predicting the binding energies of neutral,protonated,and deprotonated water clusters[J]. J Chem Theory Comput,2009,5(4):1016-1026.
[16] 孙涛,王一波.用GGA密度泛函及其长程、色散校正方法计算各类氢键的结合能[J].物理化学学报,2011,27 (11):2553-2558.
[17] GUO J J,ZHU C,HE Q Q,et al.Infrared spectra and pyrolysis of selected molecular models of coal:insight from density functional calculations[J].结构化学,2013, 32(6):863-870.
[18] FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al. Gaussian 09:revision B.01[M].Wallingford,CT: Gaussian Inc,2009.
Theoretical Study on Water-holding Properties of the Functional Group of Lignite Model Compounds
ZHANG Xiangfei1,SUN Mingjun1,GUO Juanjuan1,FENG Li2,CAO Zexing1*
(1.College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China;
2.School of Chemical Engineering and Technology,China University of Mining and Technology,Xuzhou 221116,China)
Abstract:Density functional calculations have been used to explore the interactions between the water and the functional group of lignite structural components,and the effects of water-holding capacity and local chemical environment on moisture adsorption and desorption of lignite model compounds have been discussed.An approximate thermodynamic relation has been derived based on the thermodynamic cycle to evaluate the temperature and pressure influence in dewatering process.The results indicate that the dependence of the critical dewatering temperature on pressure is not remarkable.Under elevated pressure,the desorbed water molecules may prefer in an aggregate state,which can lower the dewatering energy consumption.
Key words:density function theory calculations;lignite;water-holding functional groups;dewatering;thermodynamics values
*通信作者:zxcao@xmu.edu.cn
基金项目:国家自然科学基金(21133007);国家重点基础研究发展计划(973计划)(2012CB214900)
收稿日期:2015-04-13 录用日期:2015-06-11
doi:10.6043/j.issn.0438-0479.2016.02.002
中图分类号:O 641
文献标志码:A
文章编号:0438-0479(2016)02-0155-07
引文格式:张向飞,孙铭骏,郭娟娟,等.褐煤模型化合物中官能团持水性质的理论研究[J].厦门大学学报(自然科学版),2016, 55(2):155-161.
Citation:ZHANG X F,SUN M J,GUO J J,et al.Theoretical study on water-holding properties of the functional group of lignite model compounds[J].Journal of Xiamen University(Natural Science),2016,55(2):155-161.(in Chinese)
