石墨烯带隙的调控及其研究进展
2016-04-07王华平
蔡 乐,王华平,于 贵
中国科学院化学研究所,北京分子科学国家实验室,北京100190
石墨烯带隙的调控及其研究进展
蔡乐,王华平,于贵∗
中国科学院化学研究所,北京分子科学国家实验室,北京100190
摘要:石墨烯是一种单原子层的二维材料,因其独特的晶格结构而具备十分优越的性能,引起了科学家的广泛关注。但因其价带与导带相交于狄拉克点,导致石墨烯为没有带隙的半金属,限制了其在纳电子学器件中的应用。为了打开石墨烯的带隙,研究者们付出了巨大的努力。在石墨烯中引入带隙的方法包括量子限制法、掺杂法和对称性破缺法,它们分别是将电子限制在一维的石墨烯纳米带中、对石墨烯进行n-型或p-型掺杂以及在双层石墨烯的垂直方向施加外加电场使双层石墨烯的对称性破缺。本文着重介绍石墨烯纳米带的合成法、石墨烯掺杂的种类和打破双层石墨烯对称性的方法。
关键词:石墨烯;带隙;调控;纳米带;掺杂;双层石墨烯
*yugui@iccas.ac.cn
目录
I.石墨烯简介21
II.石墨烯纳米带22
A.切割碳纳米管法22
1.混酸切割碳纳米管法22
2.电极切割碳纳米管法23
3.金属粒子催化裂解碳纳米管法23
4.等离子体刻蚀碳纳米管法24
B.刻蚀石墨烯法24
1.等离子体刻蚀石墨烯法24
2.金属粒子辅助刻蚀石墨烯法25
3.光学刻蚀石墨烯法26
C.小分子合成法26
1.有机合成法26
2.化学气相沉积法26
III.石墨烯的掺杂27
A.吸附掺杂28
1. p-型掺杂28
2. n-型掺杂29
B.晶格掺杂29
1. n-型掺杂29
2. p-型掺杂30
IV.对称性破缺法30
V.结论与展望31
致谢31
参考文献31
I.石墨烯简介
2004年,英国曼彻斯特大学的两位科学家Geim和Novoselov通过微机械剥离法制备了石墨烯(graphene)[1],开启了二维纳米材料的新篇章。石墨烯是一种新型的二维碳材料,只有一个原子尺寸的厚度,是具有类似蜂窝状结构的原子晶体,C-C键的长度约为0.142 nm,碳原子采用sp2杂化,晶格内的三个σ键紧密相连形成稳定的六边形结构;另一个垂直于平面的pz轨道形成共轭大π键。石墨烯具有优异的电学性能,室温低载流子浓度下测得迁移率达到140000 cm2V−1S−1,中等载流子浓度下也达到40000 cm2V−1S−1以上[2],远高于传统的硅半导体材料和迁移率最高的二维半导体异质结的数值。这是由于垂直于平面的π电子离域在整个体系,有利于载流子的传输。石墨烯的电导率可达106Sm−1,面电阻为31 Ωsq−2,是已知材料中导电性能最佳的明星材料。石墨烯是集高透过率和高导电性为一体的神奇材料,石墨烯的吸光性能只与石墨烯的层数有关,与光的波长与强度无关。单层石墨烯的吸光强度为恒定的常数[3],约为2.3%,即透过率为97.7%。随着石墨烯层数的增加,透过率逐级递减。石墨烯在电学和光学方面拥有优越性能,促进了其在纳电子学器件中的应用。但是本征石墨烯是一种零带隙的半金属材料,导带和价带相交于布里渊区K(K′)点[4],能带难以打开,限制了其在构建石墨烯基电学器件中的应用。因此只有打开石墨烯的能带才能真正实现石墨烯在纳电子学领域中的应用。打开石墨烯能带常用的方法有量子限制法、掺杂法以及对称性破缺法。量子限制法是将石墨烯制备成石墨烯纳米带(Graphene nanoribbons, GNRs),由于量子效应和边缘效应使之能够有效打开能带。GNRs传承了石墨烯的许多优异性能又具有能隙。因此相比于石墨烯,GNRs在纳电子学领域具有更大的潜在实用价值。另外,对石墨烯进行原子掺杂和吸附掺杂,可以使石墨烯的费米能级在狄拉克点附近上下移动,进而可以有效打开石墨烯的能带。双层石墨烯的堆积方式常有AA型和AB型堆垛:AA型堆垛由于完全对称而无法打开能带;AB型堆垛对称性不高,分别在导带底和价带顶调控外加不同的电场使能级发生劈裂,进而打开能带[5]。下面将对打开石墨烯能带的方法进行介绍。
II.石墨烯纳米带
GNRs是一种准一维碳材料,类似于碳纳米管。限域宽度和量子效应导致GNRs具有独特的边缘效应[6]。GNRs根据碳链边缘结构的差异,可以分为锯齿型和扶手型[7]。两者呈现出不同的电子传输类型,通过紧束缚理论计算可知锯齿型石墨烯纳米带(Zigzag graphene nanoribbons)一般呈现金属性,而扶手型石墨烯纳米带(Armchair graphene nanoribbons)可能为金属性或者半导体型,且半导体型GNRs的带隙与宽度呈反比[8,9]。但是在GNRs的制备过程中,很难获得理想的边缘结构,常常会出现边缘的混乱。而边缘混乱会导致GNRs的质量下降,减小带隙与GNRs的宽度和边缘构型之间的依赖性,对GNRs的电学性质有很大影响[7]。因此,寻找合适的方法,使制备的GNRs宽度合适、边缘结构明确、原子级光滑是尤为重要的。GNRs的制备方法按前驱体材料进行分类,可分为切割碳纳米管法、刻蚀石墨烯法和小分子合成法。
A.切割碳纳米管法
GNRs与碳纳米管(Carbon Nanotubes, CNTs)有相同的晶格结构,都是准一维碳纳米材料。CNTs的发现比石墨烯和GNRs都要早,合成技术也相当成熟。因此从理论上讲,通过改变CNTs的长度和直径可以调控GNRs的尺寸,获得理想的GNRs。但实际上通过切割碳纳米管法获得的GNRs比较宽(100 nm左右),层数较多,产率较低,易形成CNTs和GNRs的混合物。切割CNTs常用的方法有混酸切割法、电极切割法、金属粒子催化裂解法和等离子体刻蚀法。
1.混酸切割碳纳米管法
James等人首次提出混酸切割法的概念[10],之后不少研究组不断优化实验条件,改进混酸切割法的不足,合成高质量的GNRs。在室温或者加热条件下(55~70◦C)用高锰酸钾氧化切割悬浮在浓硫酸中的多壁碳纳米管(MWCNTs),得到类似于氧化石墨烯的“氧化石墨烯纳米带”,然后用N2H4·H2O进行还原。得到具有平直边缘结构的GNRs,但残留有少许多壁碳纳米管的侧壁结构。GNRs长度可达4µm,宽度通常大于100 nm,厚度15~20 nm。用同样的方法处理单壁碳纳米管,但解开缠绕比较困难,容易扭曲和聚合,难以形成GNRs。且该方法只适用于化学气相沉积法制备的MWCNTs,对于激光炉制备的MWCNTs采用同样的处理方法没有发现类似的现象。高锰酸钾氧化切割CNTs制备GNRs主要是利用高锰酸钾的强氧化性将CNTs的碳碳双键氧化断开(如图1所示)。高锰酸钾氧化MWCNTs形成石墨烯锰酸酯是第一步,也是决定速率的一步(2,图1b);锰酸酯再氧化成酮,毗邻相互支撑的二酮使得β,γ-烯烃扭曲,有利于被高锰酸钾进一步氧化(3, 图1b);随着反应的进行,键角的张力使β,γ-烯烃更加活泼,氧化更加彻底(4,图1b);而氧质子的形成使酮进一步氧化成羧基,促使GNRs的边缘光滑和平直,最终二酮的出现表明CNTs完全打开形成GNRs(5,图1b)。此方法的优点在于可以大规模合成GNRs,边缘结构光滑,但GNRs较宽,带隙不够明显,缺陷较多,降低了其电学性能。Cataldo等人利用配比为1/3的HNO3/H2SO4混合液处理单壁碳纳米管[11],弥补了上述方法不能简易解开缠绕单壁碳纳米管的缺点。Higginbotham等人引入第二种弱酸(TFA,H3PO4)改进KMnO4/H2SO4体系,通过优化氧化温度和反应时间,可将GNRs的宽度缩小到100 nm以下,长度增加到5 mm以上,提高纵横比和产率[12]。虽然混酸法简单易操作,但制备的石墨烯纳米带缺陷较多,边缘含有氧化基团,因此,GNRs的质量较差,导电性和迁移率都下降很多。
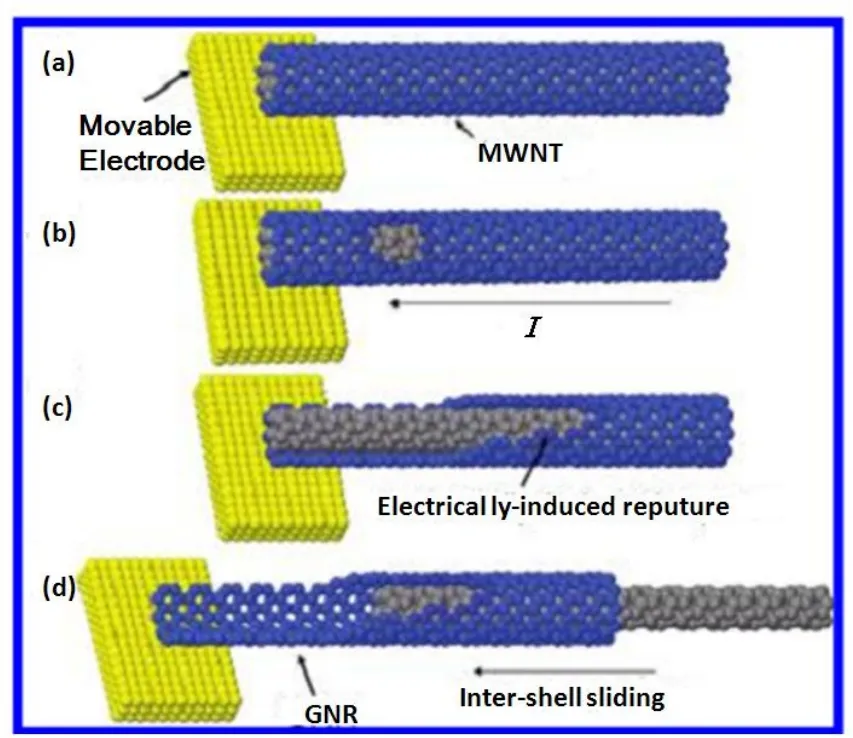
图2.电极切割法的简单示意图。(a)一端连接着移动电极的多壁碳纳米管,(b)多壁碳纳米管的局部开始裂解并逐渐扩大,(c)先驱石墨烯纳米带的形成,(d)石墨烯纳米带与碳纳米管相对位移并形成悬浮的GNRs[13]
2.电极切割碳纳米管法
混酸切割法易引入杂质,为了寻找一种无污染的切割方法,Kwanpyo Kim等人提出利用电极的移动诱发碳纳米管的外壁裂解而制备GNRs的方法[13]。在真空环境以及极高的偏压下,MWCNTs呈现出超塑性或者因为同心壁结构的破坏而容易发生结构坍塌。因此利用直流脉冲切割MWCNTs得到GNRs具有一定难度的,关键在于合理控制偏压,避免传统式灾难性的壁坍塌。电极切割法的主要过程如图2所示。首先在MWCNTs一端连接一个可移动的电极,合理控制电极的偏压使之产生适当的电流(a,图2);在电流作用下MWCNTs的外层壁开始部分裂解(b,图2);随着反应的进行,越来越多的外层壁逐渐裂解,最后形成先驱GNRs,先驱GNRs在未完全与MWCNTs的中心分离之前会吸附在MWCNTs的中心(c,图2);最后GNRs和MWCNTs的中心发生系统的移动,直到GNRs悬浮在真空中(d,图2),而且相对移动的过程在MWCNTs中心完全脱离GNRs后会自动停止。此方法制备的GNRs宽度在45 nm左右,长度约为300 nm。由于该制备GNRs的方法在真空环境下进行且没有引入化学杂质,GNRs的电学性能可以与剥离的石墨烯相媲美,如电流密度可达22 A/cm。但是此方法的合成产率低,操作困难,容易导致碳纳米管的链式坍塌,而不易大量制备GNRs。
3.金属粒子催化裂解碳纳米管法
催化法是利用金属纳米粒子催化sp2杂化的碳原子发生加氢作用而促使碳纳米管裂解,获得石墨烯纳米带的方法。Ana Laura研究了不同金属粒子对催化裂解碳纳米管的影响[14]。通常镍纳米粒子做为刻蚀剂的效果最好,获得的GNRs宽度为15~40 nm,长度为100~500 nm。通过控制金属纳米粒子的尺寸和金属纳米粒子在CNTs边缘的分布可以控制GNRs的宽度。该方法经历以下四个主要步骤:(1)将原始的MWCNTs在甲醇溶液中超声1 h,使之均匀的分散在甲醇溶液中;(2)室温下,将硅片垂直放入分散溶液中,让溶剂自然挥发。由于毛细作用,CNTs会在硅片上形成薄膜;(3)利用脉冲直流磁控溅射在带有CNTs薄膜的硅片上沉积一层2 nm的Ni薄膜;(4)在氩气氛围中,在850◦C下加热30 min,化学作用使金属纳米粒子在CNTs表面的随意移动。运动路径通常不为直线,因此CNTs的切割不是理想中的标准直线切割。金属粒子尺寸越大,切割越深,GNRs越窄。但此方法较为复杂,切割方向的随意性造成GNRs的边缘结构混乱,不平整,严重影响了GNRs的电学性质。由于初始原料为多壁碳纳米管,因此难以得到单层的GNRs。
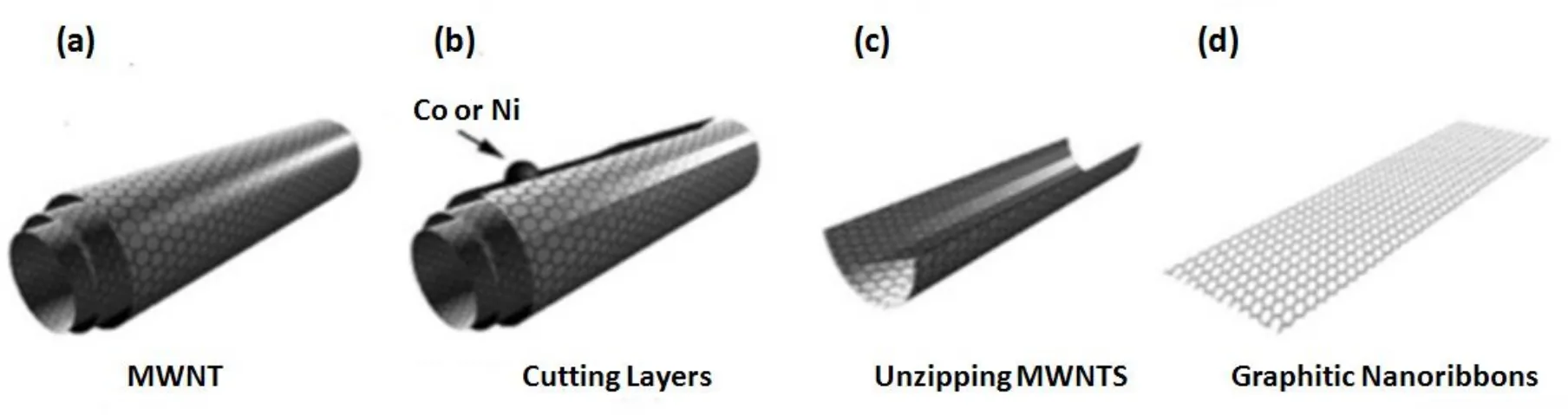
图3.金属粒子催化裂解多壁碳纳米管的示意图[14]
4.等离子体刻蚀碳纳米管法
等离子体刻蚀法制备的GNRs拥有更窄的宽度,量子限制作用更加显著,更有利于打开GNRs的带隙。等离子体刻蚀法的主要原理是使用掩膜版和等离子体刻蚀剂将未受掩膜板保护的CNTs刻蚀除去,剩下的CNTs被纵向剖开成为GNRs。Dai课题组利用聚甲基丙烯酸甲酯(PMMA)作为掩膜板和氩气等离子体刻蚀剂制备了GNRs[15],宽度10~20 nm,高度小于2 nm。他们还发现氧等离子体与碳材料反应速度快,不适合用来做刻蚀剂。首先将碳纳米管均匀分散在表面活性剂溶液中,通过毛细现象使其沉积在硅片上;然后旋涂一层300 nm厚度的PMMA作为掩膜板(b,图4),并使用KOH溶液刻蚀硅片,只留下PMMA和CNTs混合物。将混合物暴露在氩等离子体环境中,进行刻蚀反应,纵向切开碳纳米管(c,图4);最后将PMMA与GNRs混合物吸附在硅片上,再用丙酮移除PMMA,残留的PMMA可在300◦C下灼烧10 min除去,得到纯净的GNRs。实验结果表明:随着刻蚀时间的增加刻蚀程度逐渐加深,能够观察到从WMCNTs和GNRs的混合物到单一GNRs的演变过程。但是难以精准的控制刻蚀时间,所以容易得到GNRs和CNTs的混合物,并且很难得到单层石墨烯纳米带。与上节叙述的催化法比较,此方法制备的GNRs宽度更窄,更能有效的打开带隙,因此,GNRs的场效应晶体管明显体现出开关比随宽度的减小而增大,与理论一致。
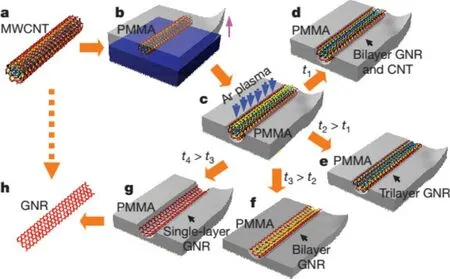
图4.等离子体切割碳纳米管的示意。(a)原始的多壁碳纳米管,(b)Si-MWCNTs-PMMA的三明治结构,(c)氩气等离子体的刻蚀(d)~(g)随着刻蚀时间的增加从MWCNTs 和GNRs的混合物到单层GNRs的过程[15]
B.刻蚀石墨烯法
纵向切割碳纳米管制备的GNRs宽度约100 nm,由于GNRs的能带与宽度成反比,GNRs的带隙通常都较小。而且由于碳纳米管的层数和切开程度深浅等问题,往往容易得到CNTs和GNRs的混合物,GNRs的层数不容易得到控制。随着研究的深入,单层石墨烯的可控生长得以实现。刻蚀石墨烯法可以改善切割碳纳米管法的缺陷,制备层数均匀的单一的GNRs。
1.等离子体刻蚀石墨烯法
等离子体刻蚀石墨烯法与刻蚀碳纳米管法的异曲同工之妙在于:利用掩膜板保护需要的石墨烯,不需要的石墨烯利用等离子体刻蚀除去就可以得到GNRs。但不同点在于掩膜板的尺寸的差异。刻蚀石墨烯法的掩膜板尺寸为纳米级别,而刻蚀碳纳米管法的掩膜板较宽。刻蚀石墨烯法常用的掩膜板一般为纳米线或者石墨烯褶皱。通过控制纳米线和石墨烯褶皱的尺寸和刻蚀时间进而调控GNRs的宽度。Bai等人利用SiO2纳米线作为掩膜版成功制备了宽度为6 nm的GNRs[16],得到的GNRs边缘光滑。将石墨烯沉积在硅片上,排列SiO2纳米线,最后用氧等离子体刻蚀掉多余的石墨烯,得到GNRs。该方法的关键在于SiO2纳米线的尺寸和沉积排列。SiO2纳米线越细,GNRs越窄。SiO2纳米线的沉积越规整越容易制备高质量GNRs,甚至可得到GNRs的阵列。刘忠范教授课题组使用纳米结构的铜箔和氩气等离子体制备了10 nm以下GNRs[17]。该方法的独特之处在于掩膜板的特殊,即未使用其他材料而使用石墨烯自身的褶皱作为掩膜板,避免了杂质的污染。该方法的具体过程如下:(1)以铜箔为基底利用CVD法制备了褶皱密集的石墨烯(a,图5);(2)使用独特的转移手段,将石墨烯的褶皱结构完整的转移至硅片上(b,图5);(3)以褶皱作为掩膜板利用氩等离子体刻蚀,除去多余的石墨烯,就可以获得GNRs(c,图5)。该方法的关键在于石墨烯褶皱的保留转移。由于石墨烯表面存在着微小的褶皱结构,利用普通的转移方法难以将石墨烯的褶皱结构完整的保留在硅片上。刘等人利用金薄膜作为保护层,成功的将褶皱结构完整的保留在硅片上。等离子体刻蚀石墨烯法可以通过调节刻蚀时间来控制GNRs的宽度,能够有效地打开带隙。

图5.以褶皱为掩膜板制备GNRs的示意图。(a)在纳米结构的铜箔上采用CVD法制备的石墨烯,(b)位于硅片上褶皱结构保留完整的石墨烯,(c)氩气等离子体刻蚀后得到GNRs[17]
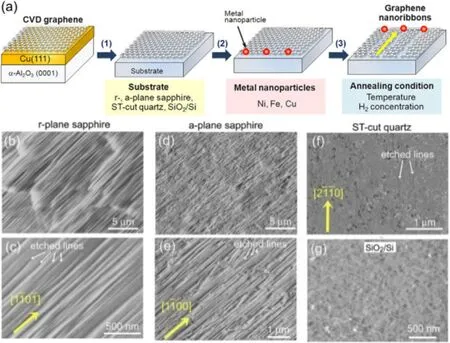
图6.在不同基底表面金属粒子对石墨烯的各向异性刻蚀。(a)金属各向异性刻蚀的示意图,(b, c)r-蓝宝石基底上的刻蚀痕迹,(d, e)a-蓝宝石基底上的石墨烯刻蚀痕迹,(f)ST-cut石英基底上的石墨烯的刻蚀痕,(g)SiO2/Si基底上的石墨烯刻蚀痕迹[18]
2.金属粒子辅助刻蚀石墨烯法
如同上节提到的金属纳米粒子刻蚀碳纳米管法,同样也可以利用金属粒子的各向异性对石墨烯进行刻蚀,制备GNRs(a,图6)。但与此不同的是利用金属粒子刻蚀石墨烯时,基底决定金属粒子移动的轨迹。Hiroki Ago小组研究了不同基底(r-, a-蓝宝石, ST-cut石英和SiO2/Si基底)对金属粒子刻蚀的影响[18]。他们发现在无定型SiO2/Si基底上进行的刻蚀是没有规律的点状,不是直线型的(g,图6);但是使用r-蓝宝石为基底时,刻蚀作用是沿着基底特殊的晶向[1¯10¯1]进行的,产生平行的刻蚀痕迹(b, c,图6);使用a-蓝宝石为基底时,在[1¯100]方向上有着线性刻蚀的痕迹,且密度大于r-蓝宝石基底(d, e,图6);而在ST-cut石英基底上,线性刻蚀的痕迹很少(f,图6)。上述结果表明使用r-, a-蓝宝石衬底有利于得到GNRs。该方法制备的石墨烯纳米带的宽度在10 nm之下,但是会残留许多金属纳米粒子,对GNRs造成了污染,降低了其电学性能。
3.光学刻蚀石墨烯法
光刻技术是集合刻蚀功能和图像复印技术,并使用先进高科技仪器的一种技术。它能可控制备尺寸一致和取向一致的GNRs。通常包括扫描隧道电子显微镜(STM)刻蚀、原子力显微镜(AFM)刻蚀和电子束刻蚀。Masubuchi等人利用AFM原位阳极氧化效应制备了GNRs[19];Shin等人利用毛细现象在AFM针尖吸附“墨水”分子构建可控的纳米模板[20],同时采用聚苯乙烯刻蚀技术辅助,制备了宽度小于25 nm的高质量高精度GNRs。电子束刻蚀技术能够制备宽度小于10 nm的GNRs,但是制备更小、更加精准尺寸的GNRs使电子束刻蚀法受到了限制。为了降低GNRs的宽度,研究者把注意力转向到STM刻蚀技术。STM刻蚀技术是一种纳米印刷技术,可实现原子尺度的制备,在水平和垂直的分辨率分别能达到0.1和0.01 nm。Levente课题组利用STM制备了宽度更小的石墨烯纳米带(2.5 nm),并且能够控制GNRs的晶格取向[21]。
C.小分子合成法
上节介绍的切割碳纳米管法和刻蚀石墨烯法都是一种由上至下的制备GNRs方法,其理念是寻找合适的“剪刀”将碳纳米管和石墨烯裁剪成GNRs。这种刻蚀方法的缺陷是GNRs边缘粗糙,污染严重,导致电学性能下降。而一种由下至上的合成法—小分子合成法被认为是不可缺少的直接制备GNRs的一种方法。小分子合成法解决了污染和缺陷等问题,使GNRs具有确定的边界结构,制备GNRs的常用小分子合成方法包括有机合成法和化学气相沉积法。
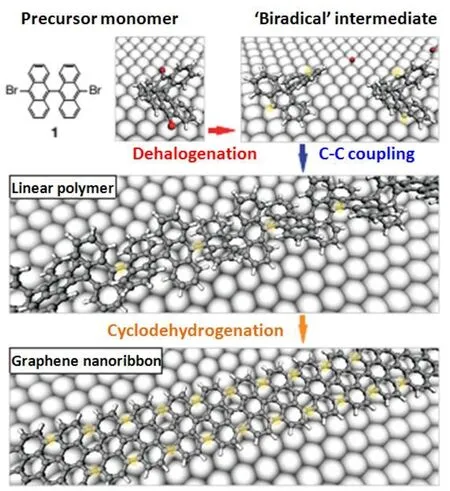
图7.有机合成法的脱卤和环化的示意图[24]
1.有机合成法
有机合成法最开始是使用多环芳烃(PAHS)制备纳米石墨烯[22,23],后来科学家们利用一维线型的聚苯类化合物做为前驱体制备了GNRs,于是有机合成法成为制备超窄GNRs的一种便捷方法。Cai等在高真空的环境下,以Au(111)为基底、二溴联二蒽(DBBA)为前驱体,通过热升华制备了只有一个原子厚度、小于10 nm宽的石墨烯纳米带[24]。该方法合成GNRs的主要过程分为两步:(1)DBBA前驱体脱去卤素取代基,形成表面稳定的聚蒽,即目标GNRs的基石结构分子;(2)在Au(111)表面的催化下,聚蒽发生脱氢环化反应,形成拓展的稳定的GNRs芳香体系(图7)。该反应的关键在于脱氢环化过程,Bjork等通过理论计算论证了Au(111)单晶对该反应的催化和两种辅助脱氢环化过程[25]。Au(111)单晶对两种脱氢过程都有催化作用,首先碳原子在Au表面有大的结合能影响脱去卤素的过程;然后Au能够吸附氢原子,降低势垒,促进脱氢环化过程。一种脱氢过程是单体间的脱氢环化,即单体在脱掉卤素取代基后不发生C-C耦合形成聚合物而是进行脱氢环化反应;另一种是聚合物之间的脱氢环化,即在单体脱掉卤素后,先发生C-C键的耦合形成长链后脱氢环化。但该有机合成GNRs的方法难以实现规模化的生产。
2.化学气相沉积法
近年来,使用化学气相沉积法(Chemical vapor deposition,CVD)制备石墨烯的技术越来越成熟,于是使用CVD法大规模制备GNRs也吸引了科学家们的注意。Toshiaki Kato等人通过对基底进行特殊处理,利用CVD法直接制备了GNRs[26],该方法制备的GNRs有着明显的带隙,约为58.5 meV,基于此石墨烯制备的场效应晶体管开关比达到104。该方法成功的关键在于纳米Ni基底的构建,运用传统的光刻技术能构建宽度为50~100 nm的纳米Ni基底。在反应的过程中,纳米Ni基底可以减小到23 nm左右。(如图8所示)该方法制备的GNRs宽度不会大于纳米Ni基底的宽度,并且转化效率(GNRs的宽度/纳米Ni基底的宽度)为45~75%。但是在CVD生长的高温环境下,金属容易团聚,会造成GNRs的断裂不连续。鲍哲楠课题组利用静电纺丝引入一维模板,成功制备了GNRs[27]。该方法的主要原理是在生长GNRs之前在基底上引入一维模板,然后以甲烷做碳源,氢气做载气使用低压CVD制备GNRs。该方法的优点是能够大规模的生产GNRs,而且当一维模板成阵列分布时可以得到GNRs阵列。魏大程利用模板CVD法实现了GNRs的可控制备[28]。主要过程是在硅基底上引入ZnS的纳米带,然后以甲烷做碳源,氢气做载气,在ZnS模板的催化下生长了GNRs,并通过控制生长时间和碳源流速实现GNRs的层数调控,但该层数调控多为少数层,难以实现GNRs的单层制备。
通过制备石墨烯纳米带打开能带是非常普遍的方法,但通常得到的带隙都比较小,且由于杂质等原因,石墨烯纳米带的电学性能受到影响,迁移率、电导率会下降。各种制备石墨烯纳米带的方法有其自己独特的优缺点具体分析比较详见表I。

图8.在纳Ni基底上CVD法制备GNRs的示意图[26]

表I.各种制备石墨烯纳米带的方法的优缺点分析
III.石墨烯的掺杂
石墨烯是一种零带隙的半金属,其价带与导带相交于布里渊区K(K′)点,具有空穴和电子两种载流子传输特性[2]。这种零带隙的结构容易受表面吸附和晶格掺杂的影响,使石墨烯的费米面在狄拉克点上下移动[29],进而引入带隙。石墨烯的这种掺杂与半导体的掺杂效应类似,一般可分为吸附掺杂和晶格掺杂;吸附掺杂是因为石墨烯具有很大的比表面积(约2600 m2g−1),在其表面容易吸附一些小分子,造成电荷的转移。晶格掺杂是指石墨烯的晶格结构中的碳原子被其它杂原子代替的掺杂。通过石墨烯掺杂引入的带隙往往大于石墨烯纳米带边缘量子化效应引入的带隙。验证石墨烯是否掺杂成功的常用方法包括构建场效应晶体管和测量拉曼光谱与X射线光电子能谱[30]。以本征石墨烯构建的场效应晶体管的转移曲线是V-型,且在V=0处有最低点。p-型掺杂石墨烯的转移曲线的最低点会向正栅压移动,n-型掺杂石墨烯反之;石墨烯拉曼光谱G峰移动能够反映石墨烯的掺杂是否成功,p-型和n-型掺杂会使G峰向高波数移动。X-射线光电子能谱通过元素键能的表征反应掺杂含量。
A.吸附掺杂
吸附掺杂的主要机制是石墨烯与掺杂剂之间的电荷转移,石墨烯的费米能级和掺杂剂的最低未占据轨道(LUMO)与最高占据轨道(HOMO)之间的相对位置决定了电荷转移的方向。p-型掺杂是因为掺杂剂的LUMO比石墨烯费米能级小,电子由石墨烯向掺杂剂转移。n-型掺杂是因为掺杂剂的HOMO比石墨烯的费米能级大,电子由掺杂剂向石墨烯转移。
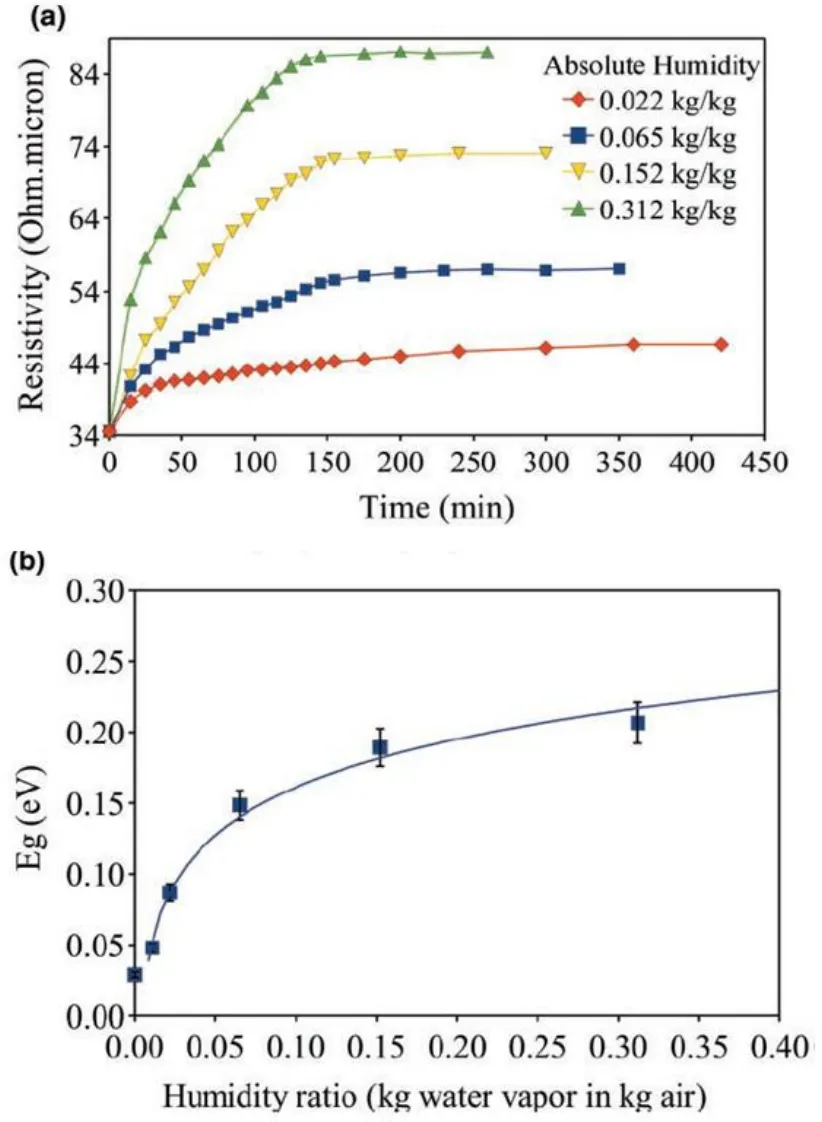
图9.(a)面电阻在不同湿度下随着时间的关系曲线,(b)绝对湿度与带隙的关系曲线[32]
1. p-型掺杂
空气中存在着许多的O2、H2O和NO2分子,这些分子具有强的吸电子效应能对石墨烯进行p-型掺杂[31]。Yavari等人研究发现在一定湿度环境下也能在石墨烯表面引入带隙[32],随着绝对湿度(每一千克干燥空气中含水的质量)的增加,带隙也将增加直至趋于稳定,最大带隙达到0.206 eV(a,图9)。同时他们也研究了面电阻和绝对湿度的关系,发现绝对湿度对面电阻的影响是缓慢的,通常达到稳定值需要几个小时;随着绝对湿度的增大,面电阻也会增大直至稳定(b,图9)。但由于这种吸附为物理吸附,其过程是可逆的,所以当石墨烯重新放置于真空中后,带隙会回到真空中的初始值(0.029 eV)。AC Crowther等人研究发现1~10层石墨烯表面吸附强氧化性分子NO2,会导致强的电子转移,形成p-型掺杂[33]。
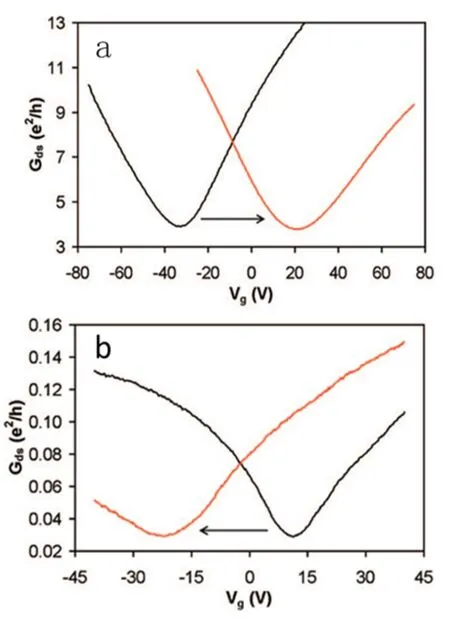
图10.(a)基于p-型掺杂石墨烯FET的转移曲线,(b)基于n-型掺杂石墨烯FET的转移曲线[38]

图11.石墨烯费米面随着o-MeO-DMBI分子浓度增加而上移[39]
以金属为催化剂合成的石墨烯常常需要转移到绝缘基底才能进行器件的构筑,而转移的过程中需要使用PMMA作为支撑材料,残留的PMMA聚合物将会对石墨烯造成p-型掺杂。Lee课题组研究含氟聚合物(CYTOP)的支撑材料对石墨烯的影响[34],发现相比于传统的PMMA支撑材料,以CYTOP做支撑材料时更容易形成深程度的p-型掺杂。其原因是在退火时或者浸泡在溶液中时,石墨烯下面的CYTOP中氟原子重新排列引入有序的偶极现象,使得电子从石墨烯转移到CYTOP。但在500◦C退火后,CYTOP发生分解,掺杂消失。石墨烯拉曼光谱的D峰和G峰向高波数位移可以证明此现象。支撑材料的影响可以对石墨烯造成p-型掺杂,转移过程的刻蚀剂也同样能够引起石墨烯的p-型掺杂。实验结果证明在转移过程中,氯化铁和过硫酸铵等刻蚀剂也会使石墨烯发生电荷转移,形成p-型掺杂[35]。

图12.固定碳氮源制备n-型掺杂石墨烯的示意图[41]
2. n-型掺杂
石墨烯在化学活性上表现为还原性,容易失去电子,因此往往容易形成p-型掺杂。要想实现将掺杂剂的电子转移到石墨烯上,不是一件容易的事情[36],因此石墨烯的n-型吸附掺杂比较少见。最早发现石墨烯有n-型掺杂特性的实例是由SiC外延生长的石墨烯,由于基底有少部分电子流向石墨烯而造成n-型掺杂[37]。随后Damon B发现当聚乙烯亚胺和重氮盐吸附在石墨烯表面时,会造成电子和空穴的不对称输运[38]。重氮盐会压制电子的输运,保留空穴的输运而导致p-型掺杂(a,图10)。而聚乙烯亚胺富含胺,是常见的电子给体容易导致n-型掺杂(b,图10)。导致这种不对称输运现象的原因是不平衡的载流子注入。强的电子给体能对石墨烯进行吸附性的n-型掺杂,Wei等人发现2-(2-甲氧苯基)-1, 3-二甲基-2, 3-双氢-1H-苯并咪唑(o-MeO-DMBI)是一种强的电子给体,当它吸附在石墨烯表面上会导致n-型掺杂[39]。随着o-MeO-DMBI浓度增加,费米面发生移动:当o-MeO-DMBI为0 mg/mL时是p-型掺杂,可能是因为石墨烯吸附了空气中的O2等气体;当o-MeO-DMBI浓度为0.01~0.1 mg/mL时,未发现掺杂现象,位于中性点,这是由于中和之前的p-型掺杂;当o-MeO-DMBI浓度大于1 mg/mL时,观察到明显的n-型掺杂现象(如图11所示)。
B.晶格掺杂
石墨烯的合成方法多种多样,常用的方法包括机械剥离法、CVD法和氧化还原法等。CVD法生长石墨烯的过程就是碳原子重新排列组合的过程,在石墨烯未形成稳定的蜂窝状结构时,很容易发生掺杂,使其它原子代替碳原子,实现石墨烯的晶格掺杂。通常来讲多电子原子易形成n-型掺杂,缺电子原子形成p-型掺杂。理论研究表明晶格掺杂可以修饰石墨烯的能级,随着掺杂浓度的增加,石墨烯带隙增大。
1. n-型掺杂
吸附掺杂容易导致石墨烯p-型掺杂是因为空气中存在大量H2O、O2等分子,所以吸附掺杂一般以p-型掺杂为主。而在晶格掺杂中,n-型掺杂的发展明显优于p-型掺杂。魏大程利用CVD法首次实现了氮掺杂石墨烯的制备[40]。主要过程是在硅基底上镀一层25 nm厚的铜薄膜做催化剂,以甲烷和氨气分别做碳源和氮源,氢气和氩气做载气,在800◦C下加热10 min制备了具有蜂窝结构的氮掺杂的石墨烯。但该方法生长的石墨烯往往都是少数层的,单层只是偶尔被发现,而且可控性不强,无法实现选择性掺杂。在此工作基础上,Zhang等人通过在金属上镶嵌氮源和碳原,制备了带隙为0.16 eV的n-型掺杂石墨烯[41],这种镶嵌碳源和氮源的优点在于可以精确控制掺杂浓度和在所希望的位点上进行掺杂以实现图案化。制备n-型掺杂的石墨烯过程如图12所示:首先通过电子束蒸发的方法在真空退火的条件下形成一个Ni(C)/B(N)/SiO2/Si基底的三明治结构,在真空退火的过程中,用硼捕获的氮原子和镍捕获的碳原子都扩散进入镍表面,在镍的催化下形成氮掺杂的石墨烯。实际上在此过程中,硼薄膜并没有参与石墨烯的掺杂,而是扮演一个完美的氮源载体。通过CVD法制备的氮掺杂的石墨烯可以用作非金属电极,有着更高的电催化活性、更长的工作稳定性和对交叉效应更强的韧性[42]。
采用电弧放电法同样可以制备氮掺杂的石墨烯。Panchakarla等人利用石墨电极的电弧放电制备了不同类型的掺杂石墨烯[43]。他们发现在H2和吡啶的存在下,石墨电极的电弧放电可以制备氮掺杂的石墨烯;在H2和NH3的存在下,石墨电极的电弧放电同样也可以制备氮掺杂的石墨烯。同时他们还发现了在吡啶的存在下,纳米金刚石可以转化为氮掺杂的石墨烯。Some等人发现除了氮原子能对石墨烯进行n-型掺杂外,磷原子同样也可以对石墨烯进行n-型掺杂[44]。
2. p-型掺杂
与n-型晶格掺杂相比,p-型晶格掺杂就像处于婴儿时期,最初报道的p-型掺杂主要以硼原子为掺杂剂。Zhu等人利用CVD法,以乙醇和硼粉作前驱体,制备了硼掺杂的石墨烯[45]。X射线光电子能谱显示石墨烯中含有硼、铁(刻蚀剂污染)和氧元素,证明了石墨烯p-型掺杂的成功。但由于乙醇和硼粉热分解的速率与硼掺杂石墨烯的合成速率不一致,因此很难控制硼掺杂石墨烯的生长。而且由于B-C键的键能小于C-C键的键能,因此合成的石墨烯热稳定性不高。Wang等人使用C6H7BO2作独立的前驱分子,同时提供碳源和硼源能够可控制备大面积、高质量的硼掺杂石墨烯[46](如图13所示)。前驱分子中的C原子和B原子在铜箔的催化下石墨化;O原子以H2O和COX的形式随着载气移除系统。而且因为铜对碳的低溶解度,硼掺杂的石墨烯薄膜大多是单层的。X-射线光电子谱证明了硼原子镶嵌在石墨烯的晶格中,所制备掺杂石墨烯的场效应晶体管迁移率达到800 cm2V−1S−1,转移曲线最低点位移到30 V附近,也证明了石墨烯p-型掺杂的成功。
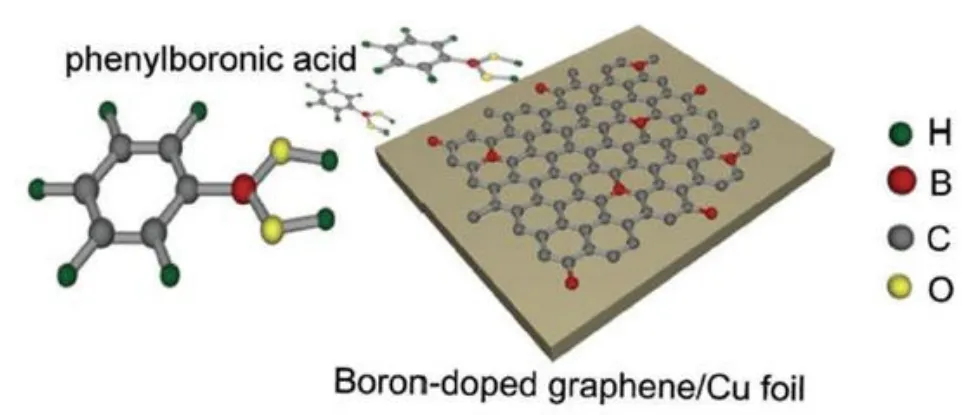
图13.以C6H7BO2为前驱体制备p-型掺杂石墨烯示意图[46]
Dai课题组研究氯等离子体、氟等离子体和氢等离子体与石墨烯的反应速率,发现氯等离子体和石墨烯反应速率比氟和氢等离子体的要慢[47],并且能实现石墨烯可控无损的p-型掺杂。之前报道的用F[48,49]、H[50]原子修饰石墨烯时,往往由于反应速率过快,容易导致石墨烯从半金属转变为绝缘体,形成石墨烷。如果改用氯等离子体,由于反应速率慢且可控,能对石墨烯进行p-型掺杂而又不损害其结构。除了氯等离子体能对石墨烯进行p-型掺杂外,Zhou等人发现氯自由基同样也可以对石墨烯进行掺杂[30,44]。掺杂后的石墨烯的拉曼光谱G峰和D峰的展宽和场效应晶体管的转移曲线最低点向正栅压移动,都能证明石墨烯p-型掺杂的成功。
石墨烯的掺杂是引入带隙最直接的方法,使费米能级移动可以形成p-型或n-型掺杂。吸附不同的掺杂分子,导致费米能级改变,产生不同的掺杂类型,稳定性也不一致,具体分析见表II。
IV.对称性破缺法
石墨烯的晶格对称性对其能带结构影响深远。除引入杂原子能导致对称性的破坏,石墨烯的堆垛结构也能造成石墨烯的晶格结构发生改变。常见的石墨烯堆垛方式有AA型堆垛和AB型堆垛。Ohta等人通过对双层石墨烯价带和导带结构的研究[51],发现基底或杂质原子等引入的偶极场可以选择性控制双层石墨烯层的载流子浓度从而轻易地调控石墨烯狄拉克点附近的能带结构。他们通过将钾原子沉积在双层石墨烯靠近真空侧的石墨烯表面上,钾原子提供价电子给上层石墨烯,而下层石墨烯未得到电子并由此产生偶极场。偶极场的存在导致双层石墨烯的对称性受损,费米能级移动从而引入能隙。Zhang等人通过对AB型堆垛的双层石墨烯施加垂直电场,成功打开了双层石墨烯的带隙,约为250 meV[52]。原始的AB型堆垛的双层石墨烯导带底和价带顶相交于狄拉克点,通过制备双栅场效应晶体管在顶栅和底栅施加不同的电压Dt和Db,可控制导带和价带中载流子浓度的差异。电压的施加会导致费米能级的移动,并且随着平均电压(1/2(Dt+Db))强度的增加,费米能级的移动越偏离平衡位置,带隙也就越大(如图14所示)。虽然这两种方法调控双层石墨烯载流子浓度差异的手段不一致,但主要原理都是使双层石墨烯的上下两层具有不同的载流子浓度,从而破坏双层石墨烯的对称性,使费米能级移动,进而引入能隙。

表II.不同掺杂类型的分析和对比

图14.带隙与平均栅压的关系曲线[52]
V.结论与展望
石墨烯由于其优越的物理和化学性质而受到人们广泛的关注。本征石墨烯是一种半金属材料,石墨烯带隙的打开是其在纳电子学领域应用的关键。石墨烯带隙为零,使得石墨烯基的场效应晶体管难以截止,即使在电中性点也存在较大电流,限制了开关比。但通过各种方法,改变石墨烯的能带结构,即引入带隙,可使石墨烯真正成为半导体材料。常用来打开带隙的方法是量子限制法、掺杂法以及对称性破缺法法。越来越多的新的方法(石墨烯量子点、石墨烯的P-N结和外加应力)也可以打开带隙,但这些的方法的原理大多与本文提到的基本方法有异曲同工之妙。石墨烯优异的电学性质源于其独特的能带结构,即导带与价带相交于狄拉克点,电子有效质量为零。而现有的方法在打开石墨烯带隙的同时破坏了狄拉克锥点,使得石墨烯的导电性能和迁移率大幅度下降。使石墨烯从优异的电导材料转变为平庸的半导体,但近期研究表明通过调制石墨烯FET沟道宽度可以平衡引入能带的和高迁移率之间的矛盾,使石墨烯具有超高迁移率的同时也能打开其带隙,约为100 meV[53]。以及在含氮种子的单晶SiC表面外延生长弯曲的石墨烯可以极大程度打开石墨烯的带隙(0.7 eV),使石墨烯保持金属性的同时拥有半导体特性[54]。石墨烯的最新研究进展表明其应用范围广泛,无论是在熔融态的玻璃表面上直接生长石墨烯[55],作为透明导电电极,还是将石墨烯应用到在隐形眼镜上[56]制作简便的红外探测仪都显示出了石墨烯优异的性能。相信在不久的将来,科学家们有望能让石墨烯走进我们的日常生活中,成为一个新的时代的划分点。
致谢
感谢国家自然科学基金(批准号:61390502)、国家重点基础研究发展计划(批准号:2013CBA01602)和中国科学院B类战略性先导科技专项(批准号:XDB12030100)项目的资助。
[1] Novoselov K S, Geim A K, Morozov S V, Jiang D, Zhang Y, Dubonos S V, Grigorieva I, Firsov A A. Science, 2004, 306(5696):666-669
[2] WangL, Meric I, Huang P Y, Gao Q, Gao, Y, Tran H, Taniguchi T, Watanabe K, Campos L M, Muller D A. Science , 2013, 342(6158):614—617
[3] Nair R R, Blake P, Grigorenko A N, Novoselov K S, Booth T J, Stauber T. Science, 2008, 320(5881):1308
[4] Son Y W, Cohen M L, Louie S G. Nature, 2006, 444(7117):347-349
[5] Oostinga J B, Heersche H B, Liu X L, Morpurgo A F, Vandersypen L M K. Nat. Mater., 2008, 7(2):151-157
[6] Dutta S, Pati S K. J. Mater. Chem., 2010, 20(38):8207-8223
[7] Haskins J, Kinaci A, Sevik C, Sevincli H, Cuniberti G, Cagin T. ACS Nano, 2011, 5(5):3779-3787
[8] Wassmann T, Seitsonen AP, Saitta AM, Lazzeri M , Mauri F. J. Am. Chem. Soc., 2010, 132(10):3440-3451
[9] Shemella P, Zhang Y, Mailman M, Ajayan P M, Nayak S K. Appl. Phys. Lett., 2007, 91(4):042101
[10] Kosynkin D V, Higginbotham A L, Sinitskii A, Lome-da J R, Dimiev A, Price B K, Tour J M. Nature, 2009, 458(7240):872-876
[11] Cataldo F, Compagnini G, Patane G, Ursini O, Angelini G, Ribic P R, Margaritondo G, Cricenti A, Palleschi G, Valentini F. Carbon, 2010, 48(9):2596-260
[12] Higginbotham A L, Kosynkin D V, Sinitskii A, Sun Z Z, Tour J M. ACS Nano, 2010, 4(4):2059-2069
[13] Kim K, Sussman A, Zettl A. ACS Nano, 2010, 4(3):1362-1366
[14] Elias A L, Botello-Mendez A R, Meneses-Rodriguez D, Gonzalez V J, Ramirez-Gonzalez D, Ci L, Munoz-Sandoval E, Ajayan P M, Terrones H, Terrones M. Nano Lett., 2009, 10(2):366-372
[15] Jiao L Y, Zhang L, Wang X R, Diankov G, Dai H J. Nature, 2009, 458(7240):877-880
[16] Bai J, Duan X, Huang Y. Nano Lett., 2009, 9(5):2083-2087
[17] Pan Z H, Liu N, Fu L, Liu Z F. J. Am. Chem. Soc., 2011, 133(44):17578-17581
[18] Ago H, Kayo Y, Solis-Fernandez P, Yoshida K, Tsuji M. Carbon, 2014, 78:339-346
[19] Masubuchi S, Arai M, Machida T. Nano Lett., 2011, 11(11):4542-4546
[20] Shin Y S, Son J Y, Jo M H, Shin Y H, Jang H M. J. Am. Chem. Soc., 2011, 133(15):5623-5625
[21] Tapaszto L, Dobrik G, Lambin P, Biro L P. Nature Nanotech., 2008, 3(7):397-401
[22] Pisula W, Feng X, M¨ullen K. Adv. Mater., 2010, 22(33):3634-3649
[23] Pisula W, Feng X, Mu╨len K. Chem. Mater., 2010, 23(3):554-567
[24] Cai J M, Ruffieux P, Jaafar R, Bieri M, Braun T, Blankenburg S, Muoth M, Seitsonen A P, Saleh M, Feng X L. Nature, 2010, 466(7305):470-473
[25] Bjo╮k J, Stafstro╩S, Hanke F J. J. Am. Chem. Soc., 2011, 133(38):14884-14887
[26] Kato T, Hatakeyama R. Nat. Nanotech., 2012, 7(10):651-656
[27] Liu N, Kim K, Hsu P C, Sokolov A N, Yap F L, Yuan H T, Xie Y W, Yan H, Cui Y, Hwang H Y. J. Am. Chem. Soc., 2014, 136(49):17284-17291
[28] Wei D C, Liu Y Q, Zhang H L, Huang L P, Wu B, Chen J Y, Yu G. J. Am. Chem. Soc., 2009, 131(31):11147-11154
[29] Xu Y X, Bai H, Lu G W, Li C, Shi G Q. J. Am. Chem. Soc., 2008, 130(18):5856-5857
[30] Zhou L, Zhou L S, Yang M M, Wu D, Liao L, Yan K, Xie Q, Liu Z R, Peng H L, Liu Z F. Small, 2013, 9(8):1388-1396
[31] Moser J, Verdaguer A, Jimenez D, Barreiro A, Bachtold A. Appl. Phys. Lett., 2008, 92(12):123507
[32] Yavari F, Kritzinger C, Gaire C, Song L, Gullapalli H, Borca-Tasciuc T, Ajayan P M, Koratkar N. Small, 2010, 6(22):2535-2538
[33] Crowther A C, Ghassaei A, Jung N, Brus L E. ACS Nano, 2012, 6(2):1865-1875
[34] Lee W H, Suk J W, Lee J, Hao Y F, Park J, Yang J W, Ha H W, Murali S, Chou H, Akinwande D. ACS Nano, 2012, 6(2):1284-1290
[35] Bae S, Kim H, Lee Y, Xu X F, Park J S, Zheng Y, Balakrishnan J, Lei T, Kim H R, Song Y I. Nat. Nanotech., 2010, 5(8):574-578
[36] Ristein J. Science, 2006, 313(5790):1057-1058
[37] Gierz I, Riedl C, Starke U, Ast C R, Kern K. Nano Lett., 2008, 8(12):4603-4607
[38] Farmer D B, Golizadeh-Mojarad R, Perebeinos V, Lin Y M, Tulevski G S, Tsang J C, Avouris P. Nano Lett., 2008, 9(1):388-392
[39] Wei P, Liu N, Lee H R, Adijanto E, Ci L J, Naab B D, Zhong J Q, Park, J, Chen, W, Cui Y. Nano Lett., 2013, 13(5):1890-1897
[40] Wei D C, Liu Y Q, Wang Y, Zhang H L, Huang L P, Yu G. Nano Lett., 2009, 9(5):1752-1758
[41] Zhang C H, Fu L, Liu N, Liu M H, Wang Y Y, Liu Z F. Adv. Mater., 2011, 23(8):1020-1024
[42] Qu L T, Liu Y, Baek J B, Dai L M. ACS Nano, 2010, 4(3):1321-1326
[43] Panchokarla L S, Subrahmanyam K S, Saha S K, Govindaraj A, Krishnamurthy H R, WaghmareU V, Rao C N R. Adv. Mater., 2009, 21(46):4726-4730
[44] Some S, Kim J, Lee K, Kulkarni A, Yoon Y, Lee S, Kim T, Lee H. Adv. Mater., 2012, 24(40):5481-5486 [45] Li X, Fan L L, Li Z, Wang KL, Zhong M L, Wei J Q, Wu D H, Zhu H W. Adv. Eng. Mater., 2012, 2(4):425-429
[46] Wang H, Zhou Y, Wu D, Liao L, Zhao S L, Peng H L, Liu Z F. Small, 2013, 9(8):1316-1320
[47] Wu J, Xie L M, Li Y G, Wang H L, Ouyang Y J, Guo J, Dai H J. J. Am. Chem. Soc., 2011, 133(49):19668-19671
[48] Robinson J T, Burgess J S, Junkermeier C E, Badescu SC, Reinecke T L, Perkins F K, Zalalutdniov M K, Baldwin J W, Culbertson J C, Sheehan P E. Nano Lett., 2010, 10(8):3001-3005
[49] Withers F, Dubois M, Savchenko A K. Phys. Rev. B, 2010, 82(7):073403
[50] Lu Y H, Wu RQ, Shen L, Yang M, Sha Z D, Cai Y Q, He P M, Feng Y P. Appl. Phys. Lett., 2009, 94(12):122111
[51] Ohta T, Bostwick A, Seyller T, Horn K, Rotenberg E. Science, 2006, 313(5789):951-954
[52] Zhang Y B, Tang T T, Girit C, Hao Z, Martin MC, Zettl A, Crommie M F, Shen Y R, Wang F. Nature, 2009, 459(7248):820-823
[53] Mohamad A, Awano Y. Appl. Phys. Express., 8(11)[54] Mihnev M T, Wang F, Liu G, Rothwell S, Cohen P I, Feldman L C, Conrad E H, Norris T B. Appl. Phys. Lett., 2015, 107(17)
[55] Chen Y, Sun J, Gao J, et al. Adv. Mater., 2015, 27(47):7839-7846
[56] Hsu A L, Herring P, Gabor N, et al. Nano Lett., 2011, 1
Modifying Bandgap of Graphene and its Recent Developments
Cai Le, Wang Hua-Ping, Yu Gui∗
Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
Graphene, a single atomic layer, is a kind of two-dimensional material, and it has aroused widespread concern. Graphene shows excellent physical and chemical properties because of its unique lattice structure. However, conduction band of graphene intersects with its valence band on the Dirac point, thus graphene exhibits a zero-band gap with linear band dispersion at the Fermi-level, limiting its application in nanoelectronic devices area. To solve the problem of the band gap of graphene, researchers have paid a tremendous effort. The way commonly used to introduce the band gap of graphene is quantum confinement, doping method, and twisted symmetry method. The ways introducing bandgap of graphene include electron limited in one-dimensional graphene nanoribbons, graphene conducted n-type or p-type doping and an applied electric field in the vertical direction in bilayer graphene to twist symmetry. Therefore, this article focuses on the preparation of graphene nanoribbons, graphene doping type, and introduction of how to twist symmetry in bilayer graphene.
Key words:graphene; bandgap; modification; nanoribbons; doping; bilayer graphene
文章编号:1000-0542(2016)02-0021-1321
中图分类号:O47
文献标识码:A
DOI:10.13725/j.cnki.pip.2016.01.002
Received date:2016-01-04
