A Bipartite Network-based Method for Prediction of Long Non-coding RNA–protein Interactions
2016-03-31MengquGeAoLiMinghuiWang
Mengqu Ge, Ao Li, Minghui Wang
A Bipartite Network-based Method for Prediction of Long Non-coding RNA–protein Interactions
Mengqu Ge1,a, Ao Li1,2,*,b, Minghui Wang1,2,c
1School of Information Science and Technology, University of Science and Technology of China, Hefei 230027, China
2Centers for Biomedical Engineering, University of Science and Technology of China, Hefei 230027, China
Received 12 November 2015; revised 4 January 2016; accepted 6 January 2016 Available online 22 February 2016
Handled by Zhihua Zhang
E-mail: aoli@ustc.edu.cn (Li A).
aORCID: 0000-0001-5185-7045.
bORCID: 0000-0001-9910-8967.
cORCID: 0000-0002-5788-894X.
Peer review under responsibility of Beijing Institute of Genomics,
Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.01.004
1672-0229ⓒ2016 The Authors. Production and hosting by Elsevier B.V. on behalf of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
KEYWORDS
lncRNA;
Protein;
Interaction;
Bipartite network; Propagation
Abstract As one large class of non-coding RNAs (ncRNAs), long ncRNAs (lncRNAs) have gained considerable attention in recent years. Mutations and dysfunction of lncRNAs have been implicated in human disorders. Many lncRNAs exert their effects through interactions with the corresponding RNA-binding proteins. Several computational approaches have been developed, but only few are able to perform the prediction of these interactions from a network-based point of view. Here, we introduce a computational method named lncRNA–protein bipartite network inference (LPBNI). LPBNI aims to identify potential lncRNA–interacting proteins, by making full use of the known lncRNA–protein interactions. Leave-one-out cross validation (LOOCV) test shows that LPBNI significantly outperforms other network-based methods, including random walk (RWR) and protein-based collaborative filtering (ProCF). Furthermore, a case study was performed to demonstrate the performance of LPBNI using real data in predicting potential lncRNA–interacting proteins.
Introduction
An increasing number of studies show that approximately 2% of the whole mammalian genome represents proteincoding genes, whereas the majority of the genome consists of non-coding RNA (ncRNA) genes. ncRNAs had long been regarded as transcriptional noise, but recent investigations demonstrate that ncRNAs play an important role in the regulation of diverse biological processes [1–5]. Long ncRNAs (lncRNAs), which consist of more than 200 nucleotides, constitute a large class of ncRNAs [6–7]. In the past several years, the number of identified lncRNAs has been increasing sharply because of the development of both bioinformatics tools and experimental techniques. Functional studies of lncRNAs show that mutated and dysfunctional lncRNAs are implicated in a range of cellular processes [8–12] and human diseases, ranging from neurodegeneration to cancer [13–18]. Although some lncRNAs, e.g., Xist [19] andMALAT1 [20], have been well studied, the functions of most lncRNAs remain unclear. Usually lncRNAs function through interacting with RNA-binding proteins (RBPs) [21–24]. Therefore, it is important to predict the potential lncRNA–protein interactions, in order to study the complex function of lncRNAs.
Since the experimental identification of lncRNA–protein interactions remains costly, developing effective predictive approaches becomes essential. Recently, several computational methods have been reported for predicting potential lncRNA–protein interactions. For instance, Bellucci et al. developed catRAPID in 2011 [25] by taking into account secondary structure, hydrogen bonds, and van der Waals forces between lncRNAs and proteins. Next, Muppirala et al. [26] introduced a method named RPISeq, using only sequence information of lncRNAs and proteins. Support vector machine (SVM) classifiers [27] and random forest (RF) [28] are used to predict RBPs. In 2013, Lu et al. [29] developed a novel approach, named lncPro, which uses secondary structure, hydrogen bond, van der Waals force features, and yields the prediction score using Fisher’s linear discriminate method. Later on, an approach named RPI-Pred was developed by Suresh et al. [30], they trained SVM-based approach, by extracting sequence and high-order 3D structure features of lncRNAs and proteins.
All the aforementioned methods are based on the biological characteristics of ncRNAs and proteins. CatRAPID and lncPro combined sequence and structural features of lncRNAs and proteins. RPISeq was based on sequence features. RPI-Pred used the high-order structure features of lncRNAs and proteins. However some studies show that lncRNAs generally exhibit low sequence conservation [1], which may make it difficult to predict interactions based on the intrinsic properties of lncRNAs. Biological networkbased methods are widely used in many types of studies, such as disease gene prioritization [31] and drug-target interaction prediction [32]. The development of bioinformatics technologies such as CLIP-seq and cross-linking immunoprecipitation, has enabled us to construct lncRNA–protein interaction networks. We introduce here a novel computational method, lncRNA–protein bipartite network inference (LPBNI), for the prediction of lncRNA–protein interactions. LPBNI identifies novel lncRNA–protein pairs by efficiently using the lncRNA–protein bipartite network. In order to evaluate the performance of the proposed method, we compared LPBNI with other network-based methods, including random walk (RWR) [31] and protein-based collaborative filtering (ProCF) [33]. RWR [31] has been used to predict genes associated with potential diseases. ProCF is derived from the recommendation algorithms, similar to the item-based collaborative filtering method [33]. The performance evaluation is based on leave-one-out cross validation (LOOCV) of the known lncRNA–protein interactions extracted from NPInter [34]. To further demonstrate the effectiveness of lncRNA–protein bipartite network, six lncRNAs were used to evaluate the performance of LPBNI in comparison with the existing methods, lncPro [29] and RPISeq [26]. These evaluation tests demonstrated that LPBNI outperforms the other methods significantly. In a case study, several potential interactions between lncRNAs and proteins identified by LPBNI were well supported by starBase [35], indicating the superior predictive ability of LPBNI.
Results
Performance comparison with other network-based methods on lncRNA–protein interactions prediction We compared the performance of LPBNI with RWR [31] and ProCF [33]. ProCF is based on the idea that if a protein interacts with an lncRNA, similar proteins will be recommended as interacting with this lncRNA∑. The linkage between piand ljcan be defined as:where S(p;p) Pik is the similarity between proteins piand pk. Here, we used cosine vector similarity to measure the similarity of proteins:where d(i) and d(k) are the degrees of proteins i and k, respectively.
We extracted 4870 lncRNA–protein interactions from NPInter 2.0 [34] (see‘Data collection and preprocessing”for detail). In LPBNI, for one node, at least two interactions are required to perform LOOCV. Therefore, the nodes that have only one link are not considered in the performance evaluation, so we further get 4796 lncRNA–protein interactions which match that condition, and this dataset is taken as‘gold standard’data in the LOOCV test. The receiver operating characteristic (ROC) curves and the area under the curve (AUC) obtained using these methods are shown in Figure 1. It is obvious that LPBNI shows the highest true positive rate (TPR) at each false positive rate (FPR). In addition, the AUC value of LPBNI is 0.878 (Table 1), which is higher than that obtained using RWR (0.765) and ProCF (0.738), respectively. These data suggest that LPBNI has a better predictive ability compared with RWR and ProCF. To validate the reliability of LPBNI, we compared the sensitivity, accuracy, precision, and Matthew’s correlation coefficient (MCC) of LPBNI, RWR, and ProCF with specificities of 99.0% and 95.0%, respectively. As shown in Table 1, with specificity of 99.0%, sensitivity, accuracy, precision, and MCC of LPBNI are all higher than that with RWR and ProCF. When specificity was reduced to 95.0%, sensitivity and MCC increased for all three methods, with decreased precision, although the accuracy remained comparable. However, LPBNI still showed a higher performance in terms of sensitivity, accuracy, precision, and MCC, compared to RWR and ProCF.
Thefoldenrichmentisalsousedtoevaluatetheperformance of the proposed method, which can be defined as: N/2/n [37], where N represents the number of candidate proteins, and n is the ranking of the tested protein among the candidate proteins for the evaluation. Based on the formula, the average fold enrichments are 4.007, 3.590, and 1.653 for LPBNI, RWR, and ProCF, respectively. These data suggest that LPBNI outperforms the other methods in identifying lncRNA–related proteins with a higher rank. Table 2 shows the number of lncRNA–protein interactions that were correctly retrieved at 5%, 10%, 15%, 20%, and 50% of all the prediction results, respectively. Among 4796 true interactions between lncRNAs and proteins, LPBNI achieves a higher retrieval compared with RWR and ProCF, at each of the investigated percentiles. The biggestdifferencewasobservedfor5%,where LPBNIrecovered 579 interactions successfully, and only 410 and 116 interactions were retrieved using RWR and ProCF, respectively.
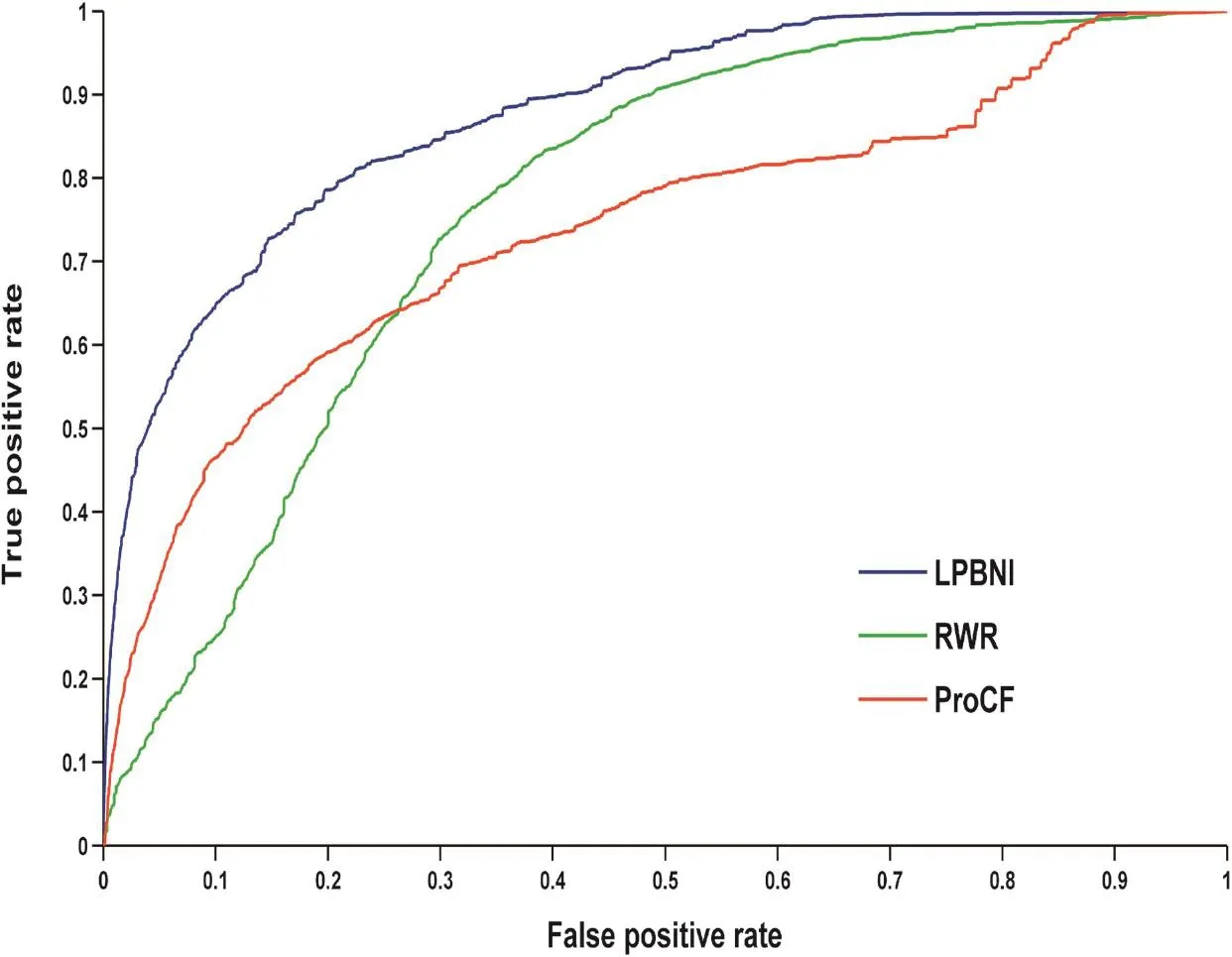
Figure 1 Performance comparison of different methods using ROC curves in predicting lncRNA–protein interactionsShown in the plot is the ROC for the whole dataset using LPBNI (blue, AUC: 0.878), RWR (green, AUC: 0.765), and ProCF (red, AUC: 0.738), respectively. LOOCV is implemented and 4796 known lncRNA–protein interactions are used as gold standard dataset. ROC, receiver operating characteristic; LPBNI, lncRNA–protein bipartite network inference; RWR, random walk; ProCF, protein-based collaborative filtering; AUC, area under this curve. LOOCV, leave-one-out cross validation.
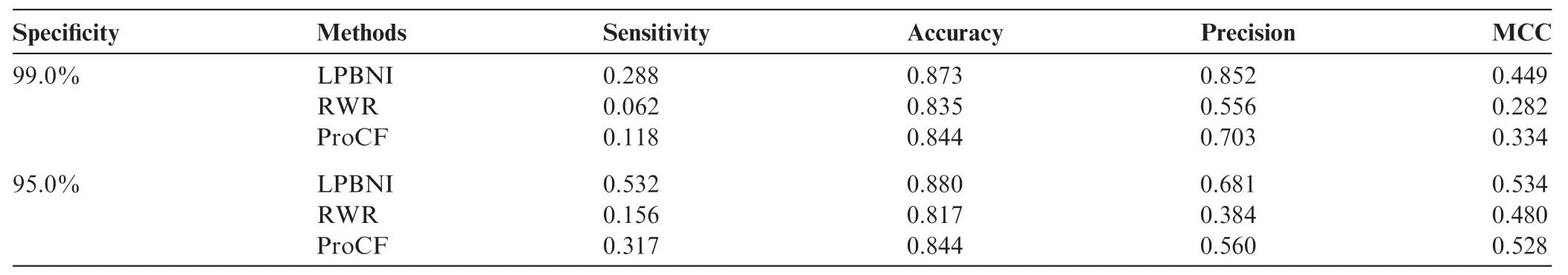
Table 1 Performance comparison of different methods with specificities of 99.0% and 95.0% in predicting lncRNA–protein interactions
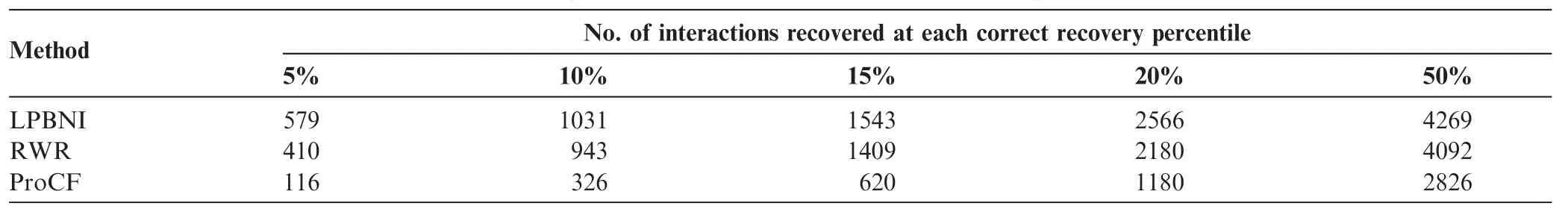
Table 2 Number of interactions that are correctly recovered from 69 true interactions using different methods
Furthermore, 10-fold cross validation was applied, in order to conduct a comprehensive performance evaluation of LPBNI in predicting lncRNA–protein interactions. All lncRNA–protein interactions (4796) are randomly divided into 10 equal portions.Eachportioninturnwasleftoutasatestsample,whilethe remaining ones were treated as training sets. During cross validation, some nodes are separated into the test sample and the corresponding links cannot be predicted by LPBIN; therefore, those links were not considered in the process of evaluation. The ROC curves for LPBNI, RWR, and ProCF are shown in
Figure S1, supporting the superior performance of LPBNI as well. Taken together, these analyses demonstrate the power of LPBNI in the prediction of lncRNA–protein interactions.
Comparison with the existing methods in predicting lncRNA–protein interactions
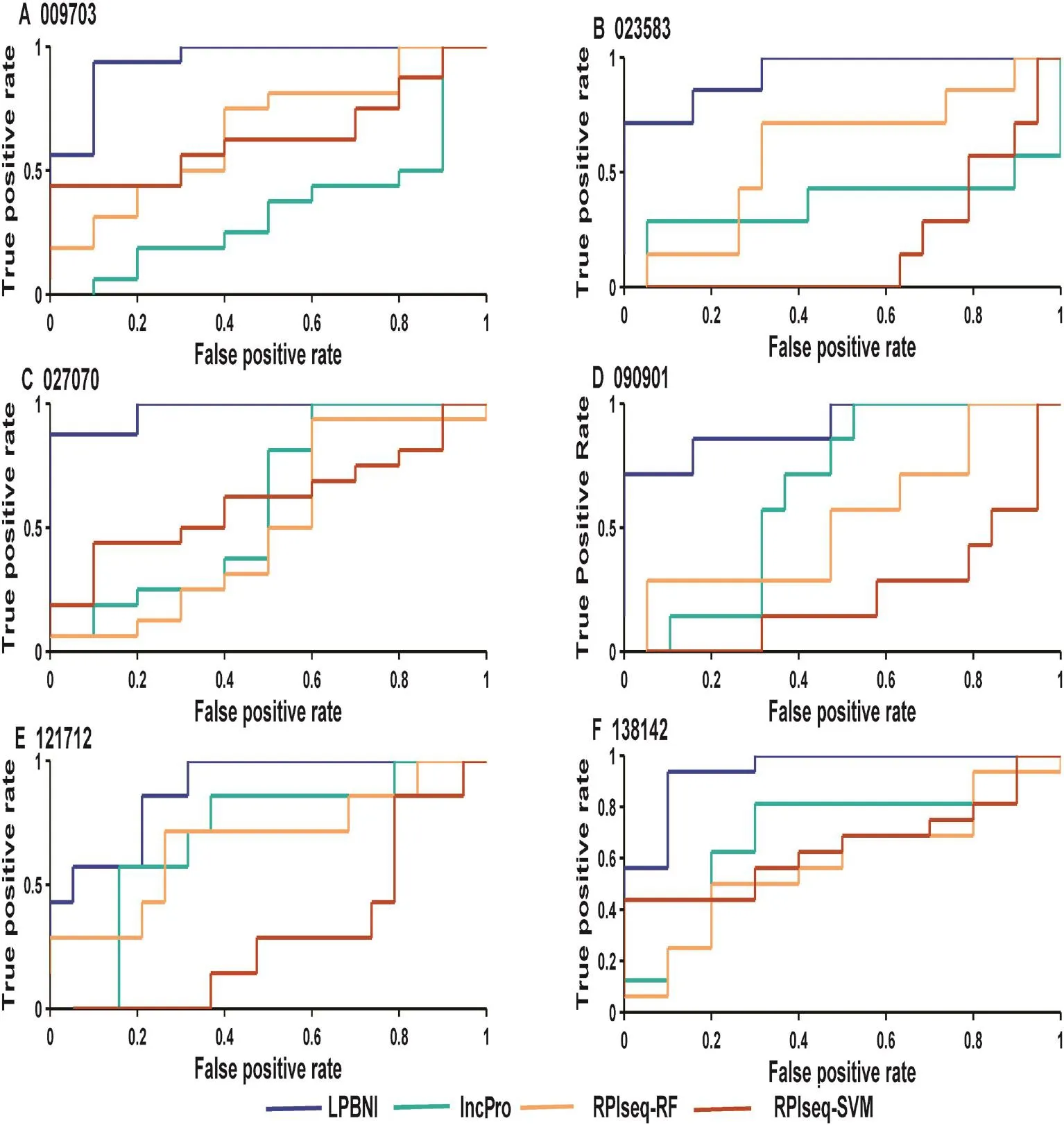
Figure 2 Performance comparison in terms of ROC curves for six lncRNAs testedShown in the plot is the ROC curves for 6 lncRNAs (A–F) using LPBNI (blue), lncPro (cyan), RPISeq-RF (yellow), and RPISeq-SVM (brown), respectively. For each lncRNA, LOOCV is performed and the corresponding lncRNA–protein interactions are used as gold standard dataset. The lncRNA IDs have been abbreviated without the‘NONHSAT”prefix, e.g.,‘009703”represents the lncRNA ‘NONHSAT009703”. ROC, receiver operating characteristic.
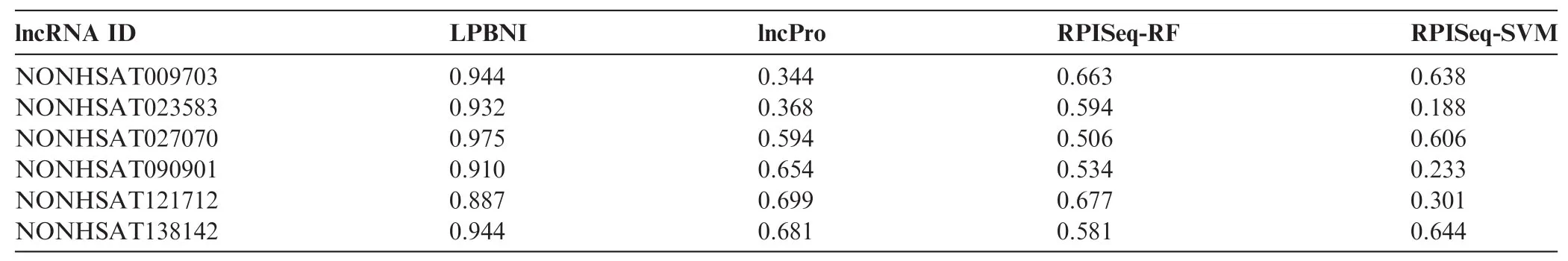
Table 3 AUC comparison of different methods for six lncRNAs
In order to further evaluate the performance of the proposed method in predicting lncRNA–protein interactions, we compared LPBNI with lncPro [29] and RPISeq [26]. RF or SVM can be used in RPISeq [26] for the prediction of lncRNA–protein interactions, which correspond to RPISeq RF and RPI-SVM here. We performed the evaluation of these methods using six lncRNAs, including NONHSAT009703, NONHSAT023583, NONHSAT027070, NONHSAT090901, NONHSAT121712, and NONHSAT138142, which we ran domly selected from the lncRNAs set and present 69 lncRNA–protein interactions. The ROC curves for predicting lncRNA–protein interactions are shown in Figure 2. For each lncRNA, the corresponding ROC curve of LPBNI is significantly higher compared with the results obtained by lncPro, RPISeq-RF, and RPISeq-SVM. As presented in Table 3, the average AUC of LPBNI is 0.375, 0.339, and 0.497 higher than lncPro, RPISeq-RF, and RPISeq-SVM, respectively. Thecomparisons among LPBNI, lncPro, and RPISeq, in terms of sensitivity and specificity, are listed in Table S1. Taking NONHSAT138142 (RP6-24A23.7) as an example, the AUC value of LPBNI was shown to be 0.944, which is 0.263, 0.363, and 0.300 higher compared with lncPro, RPISeq-RF, and RPISeq-SVM, respectively. For specificity of 95.0%, the sensitivity of LPBNI was 0.563, which is 0.438, 0.500, and 0.125 higher compared with lncPro, RPISeq-RF, and RPISeq-SVM, respectively.
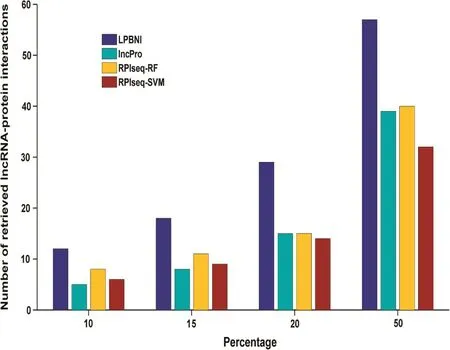
Figure 3 Number of interactions that are correctly recovered from 4796 true interactions in different percentiles4796 known lncRNA–protein interactions are taken as gold standard dataset. 10%, 15%, 20%, and 50% of all the prediction results are taken as different percentiles. The number of lncRNA–protein interactions that are correctly retrieved at different percentiles indicated the rank distribution of the lncRNA–related proteins, the higher the number in each percentile, the better the performance of the method.
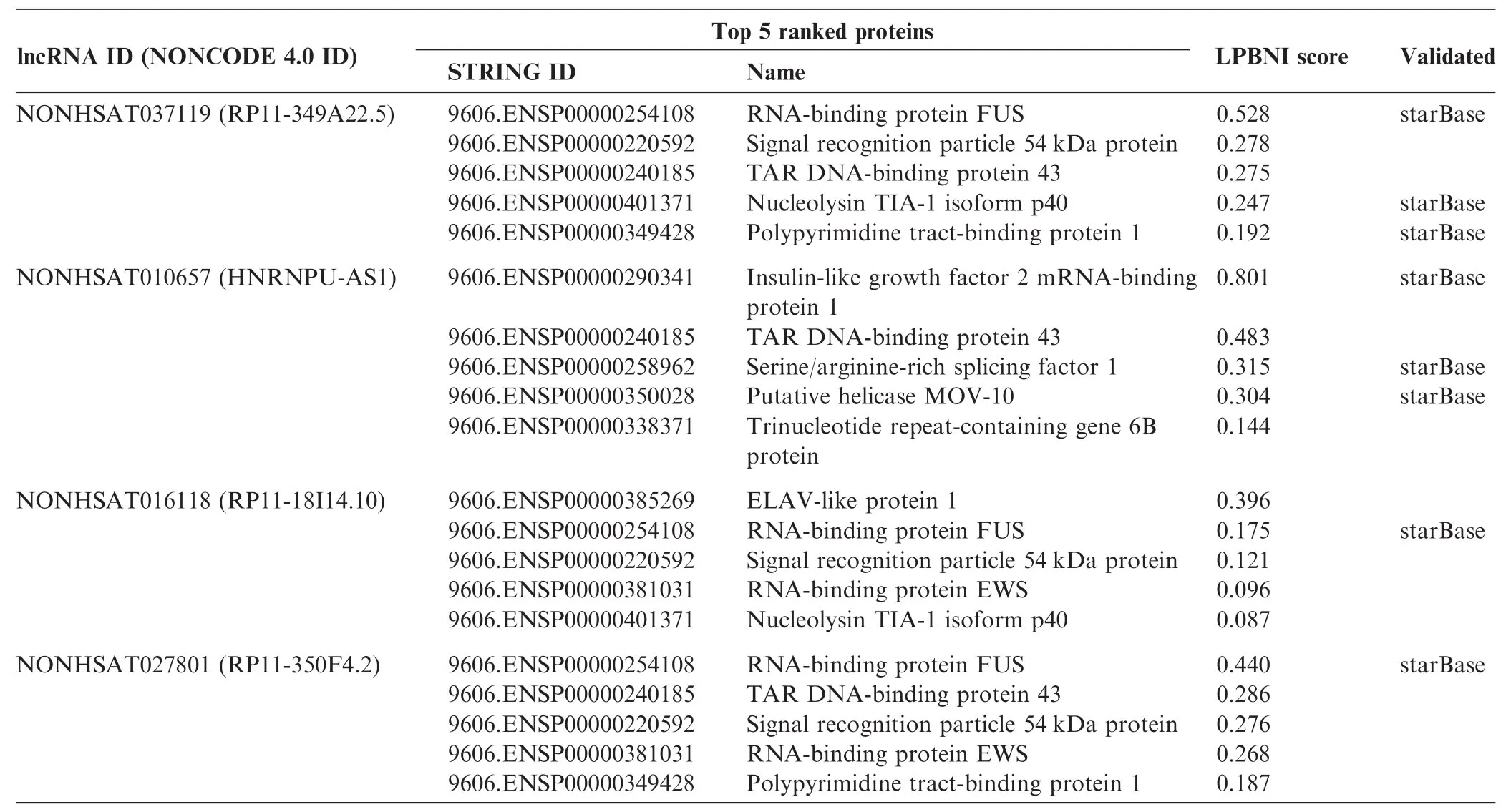
Table 4 Top 5 ranked candidate proteins for four selected lncRNAs
As aforementioned, there are 69 lncRNA–protein interaction pairs for these six lncRNAs. We next examined the num-bers of lncRNA–protein interactions that were correctly recovered with respect to different percentiles [32] of all the prediction results. As shown in Figure 3, LPBNI had the best performance for every percentile tested. Top-ranked results are of great importance, due to the low occurrence of false positive results. When looking at the top 10% of the results, 12 of 69 lncRNA–protein interactions were correctly retrieved by LPBNI, whereas only five, eight, and six interactions were correctly retrieved by lncPro, RPISeq-RF, and RPISeq-SVM, respectively. As for top 50%, LPBNI correctly recovers 57 lncRNA–protein interactions, which represent 18, 17, and 25 interactions more than lncPro, RPISeq-RF, and RPISeq-SVM, respectively. These comparisons indicate that LPBNI outperforms lncPro and RPISeq in the prediction of lncRNA–protein interactions.
Prediction of novel lncRNA–protein interactions
Following the validation of the superior performance of LPBNI using LOOCV, we applied LPBNI onto the 4796 known lncRNA–protein interactions downloaded from NPInter [34], which includes 1113 lncRNAs and 26 proteins to predict novel lncRNA–protein interactions. For each lncRNA, all the collected proteins were ranked according to the scores calculated by LPBNI, and the top five proteins are considered potential lncRNA–interacting proteins. We present here the results for four lncRNAs, which include NONHSAT037119 (RP11-349A22.5), NONHSAT010657 (HNRNPU-AS1), NONHSAT016118 (RP11-18I14.10), and NONHSAT027801 (RP11-350F4.2). Top five proteins and the corresponding scores for these lncRNAs are presented in Table 4. We searched other databases such as starBase [35] and lncRNome [36], and found that some top ranked proteins that are predicted to interact with these lncRNAs are supported by star-Base [35], which is designed to decipher miRNA-target interactions and protein-RNA interactions. As shown in Table 4 and 9606.ENSP00000254108 (RNA-binding protein FUS), 9606.ENSP00000401371 (Nucleolysin TIA-1 isoform p40), and 9606.ENSP00000349428 (Polypyrimidine tractbinding protein 1) are predicted to interact with RP11-349A22.5. 9606. ENSP00000290341 (Insulin-like growth factor 2 mRNA-binding protein 1), and 9606.ENSP00000258962 (Serine/arginine-rich splicing factor 1), and 9606. ENSP00000350028 (Putative helicase MOV-10) are predicted to interact with HNRNPU-AS1. FUS is predicted to interact with RP11-18I14.10 and RP11-350F4.2. These predictions were all confirmed by starBase [35].

Table 5 Top candidate proteins predicted by LPBNI with support by starBase and their ranks predicted using different methods
Furthermore, we extracted the predictions confirmed by starBase and compared their ranks predicted by LPBNI, lncPro, RPISeq-RF, and RPI-SVM (Table 5). The results showed that for these lncRNAs, there exist large differences in the ranks of most of the candidate proteins predicted by LPBNI, lncPro, RPISeq-RF, and RPI-SVM. Despite these great variations, candidate proteins are consistently ranked higher by LPBNI relative to the other three methods. For instance, for lncRNA RP11-350F4.2, FUS is ranked first by LPBNI, but it is ranked 12th, 15th, and 24th by lncPro, RPISeq-RF, and RPI-SVM, respectively. The results above show that LPBNI can identify potential lncRNA–interacting proteins as top candidates, implying that the use of LPBNIis a very effective way to predict novel lncRNA–protein interactions.
Discussion
In this study, we proposed and tested a novel computational method, LPBNI, for the prediction of potential lncRNA–protein interactions. We constructed an lncRNA–protein bipartite network, using the information about lncRNA–protein interactions, lncRNA and proteins are connected if they were known to interact with each other. Following this, two-step propagation was carried out in the bipartite network to score and rank candidate proteins for each lncRNA. The proposed method has some important features. Firstly, LPBNI uses only the network constructed based on the known lncRNA–protein interactions to perform this prediction. Secondly, with an increasing degree of a node, less information is assigned to its direct neighbors. Finally, the propagation matrix is not symmetrical. The results of comparisons between LPBNI and other network-based methods show that LPBNI has higher AUC, compared to RWR and ProCF. In order to further evaluate the performance of the proposed method, we compare LPBNI with the existing methods for lncRNA–protein pair prediction and obtain consistently higher ROC curves using LPBNI in relative to lncPro, RPISeq-RF, and RPISeq-SVM for the six lncRNAs tested. All the comparisons show that our method can effectively predict interactions between lncRNAs and proteins, largely by taking advantage of lncRNA–protein interaction network. The case study shows further that LPBNI is powerful not only for the recovery of known lncRNA–protein interactions, but also for the prediction of potential candidate proteins. This suggests that LPBNI may be a useful tool for predicting candidate lncRNA–interacting proteins that could be subjected to further experimental investigation for potential functional studies.
Despite the efficiency of LPBNI in the prediction of the candidate proteins for interacting with lncRNAs, some limitations exist. Firstly, LPBNI can only be implemented for a bipartite network, in which each node has at least two links. Since LPBNI only uses the prior information about the known lncRNA–protein interactions, LPBNI cannot predict candidate proteins if there is no information about the lncRNAs in the training set. This limitation may be addressed by extending the bipartite network to a bipartite network model based on lncRNA/protein functional domains or by adding the expression profile of lncRNAs [38]. Secondly, some proteins interact with a lot of lncRNAs, which may tend to get more information during the procedure of information propagation, and in consequence have higher scores in the prediction. Finally, the shortage of known lncRNA–protein interactions limits the further analysis of lncRNA mechanisms in a larger network, which may be addressed by a rapid increase in lncRNA datasets.
Conclusion
The prediction of lncRNA–protein interactions is extremely important for the studies of the complex function of lncRNAs. Existing methods are using the sequence information of lncRNAs and proteins, but in this study, we introduce a network-based method, LPBNI, which takes full advantage of the information about the known lncRNA–protein interactions. We performed the evaluation and case study of this method, which further demonstrate its superior performance.
Materials and methods
Data collection and preprocessing
7576 ncRNA–protein interactions were downloaded from the NPInter 2.0 database [34] (http://www.bioinfo.org/NPInter/) in November, 2013, with the restriction of type‘NONCODE”and organism‘Homo sapiens”. Furthermore, we extracted 2380 lncRNAs from a human lncRNA dataset downloaded from NONCODE 4.0 database [39], and converted the IDs of lncRNAs and proteins, into NONCODE 4.0 IDs and string IDs, separately. Finally, we got 4870 lncRNA–protein interactions, including 2380 lncRNAs and 106 proteins.
The lncRNA–protein bipartite network
The lncRNA–protein interaction network can be described as a graph G(L,P,E), in which L = {l1,l2,...,ln} is defined as lncRNA set, P = {p1,p2,...,pm} is defined as the protein set, and E = {ei,j|Li∈L,Pj∈P} is the edge set, where ei,jrepresents the edge connecting the nodes piand lj. A = {ai,j|i∈P,j∈L} represents the adjacent matrix, where aij= 1 if piinteracts with lj, otherwise aij= 0. For lncRNA lj, positive samples referred to the proteins that are known to interact with lj, and the remaining proteins were considered negative samples. A simple illustration of lncRNA–protein bipartite network construction is shown in Figure 4. Finally, the network was constructed and a propagation method was applied to compute the interaction score.
LPBNI method
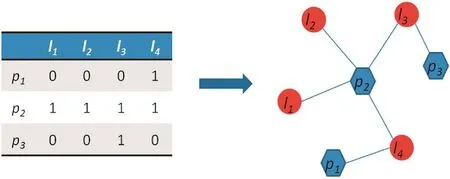
Figure 4 Example of the lncRNA–protein interaction bipartite networkThe left panel is the adjacent matrix A extracted from the known lncRNA–protein interactions, where aij= 1 if piinteracts with lj, otherwise aij= 0. The right panel is the bipartite network constructed based on the adjacent matrix. The red circles represent lncRNAs and blue hexagons represent proteins, while the known lncRNA–protein interactions are represented in blue lines.
The propagation process of LPBNI was derived from the recommendation algorithms proposed by Zhou et al. [40], and developed for the prediction of lncRNA–protein interactions. The proposed method makes full use of the information aboutthe known lncRNA–protein interactions, and scores candidate proteins for each lncRNA. We classified the nodes of lncRNA–protein interaction network into two different sets, named P and L as aforementioned and only the connections between different sets were allowed. LPBNI procedure is illustrated in Figure 5. For example, if the initial information of three proteins was 1, 1, and 0, we first propagated information from proteins to the corresponding lncRNAs. Afterward, the information was allocated from lncRNAs back to proteins. Since the network is unweighted, the information in a protein is equally propagated to its direct neighbors in the lncRNA set, and vice versa. The propagation of information after each step is shown in Figure 5B and C, respectively. This two-step propagation can be represented as:
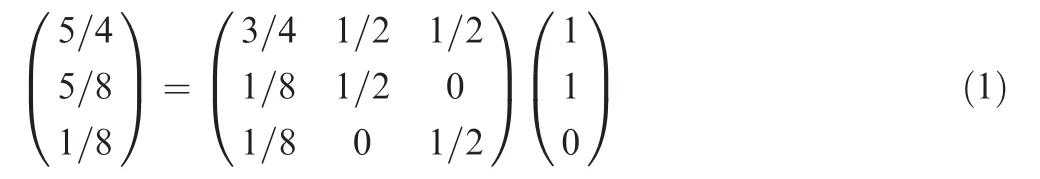
Here, 3×3 matrix represents the propagation matrix. Following the two-step propagation on the bipartite network, the final information of these three proteins becomes 5/4, 5/8, and 1/8, respectively.
For the lncRNA–protein interaction bipartite network G(L, P,E), W is defined as the propagation matrix, where wikrepresents the information transferred from pkto pinode, and can be interpreted as the importance of pifor node pk. For an lncRNA lj, we define S0(i) = sij, i∈{1,2...m} as the initial
information of protein P, sij= 1 if piinteracts with lj, otherwise sij= 0. SL(lj), j∈{1,2...n} represents the score on ljafter the first step of information propagation, which can be calculated as:

where d(pi)=∑nj=0aijis the number of lncRNAs that interact with pi.
In the second step, all the information in L propagates back to P. SF(pi) is defined as the final information of protein pi, representing the interaction score of protein piwith lj. SFcan be defined as
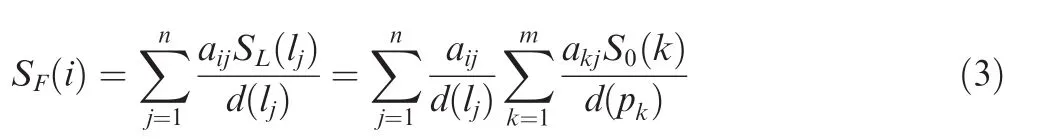
where d(lj)is the number of proteins that interact with lj.
The final information SFcan be defined in the matrix form as

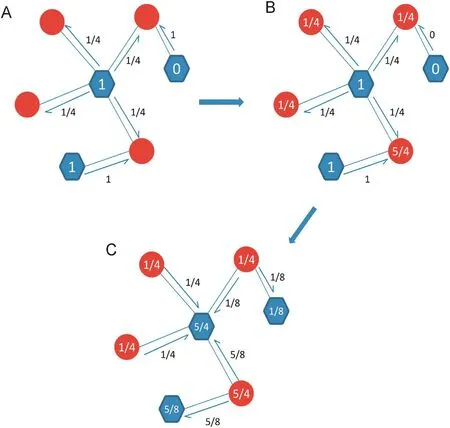
Figure 5 Illustration of the LPBNI in bipartite networkA. The process of the initial information propagated from proteins to their direct neighbor lncRNAs. For example, the initial information of three proteins is 1, 1 and 0, respectively. B. The score on red circles is the information of each lncRNA received from proteins. C. The process of the information propagated from lncRNAs back to proteins. The score on blue hexagon in panel C is the final information of each protein after the two-step propagation. The red circles represent lncRNAs and the blue hexagons represent proteins.
lncRNA–protein interaction network. Eq. (3) can be represented as:

Following the calculations, the proteins were ranked for ljby the final score SF. All of the candidate proteins are listed in a descending order, and highly-ranked proteins are considered to interact with lncRNA lj. The data and source code are freely available at https://github.com/USTC-HILAB.
Experimental design
LOOCV was performed on the lncRNA–protein interaction network for performance evaluation of the proposed method. In this process, each lncRNA–protein pair was left out in turn as a test sample, by setting the corresponding value in the adjacent matrix A to 0. The performance of LPBNI was estimated by the success rate it achieves in recovering the known lncRNA–protein interactions. In order to assess the performance of LPBNI, we plotted the ROC curves, and compared the AUC values obtained using LPBNI, RWR, and ProCF. Additionally, we computed Sp, Sn, Acc, Pre, and MCC values. The propagation matrix W presented in this paper relies on the adjacent matrix A of the bipartite network. When LOOCV was implemented, we obtained different W values, due to the change of A values in each step of LOOCV. Therefore, W value was recalculated for each lncRNA–protein pair that was left out as test sample. Furthermore, during LOOCV process, no information was propagated on the nodes with less than two links, and these nodes were not considered during the performance evaluation.
Authors’contributions
MG participated in the downloading and preprocessing of the datasets, carried out the design and performance evaluation of LPBNI in predicting lncRNA–protein interactions. AL conceived of the project and helped with the study design. MW was involved in data analysis. MG drafted the manuscript with the help of AL and MW. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (Grant Nos. 61571414 and 61471331).
Supplementary material
Supplementary material associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j. gpb.2016.01.004.
References
[1] Pang KC, Frith MC, Mattick JS. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet 2006;22:1–5.
[2] Koerner MV, Pauler FM, Huang R, Barlow DP. The function of non-coding RNAs in genomic imprinting. Development 2009;136:1771–83.
[3] Laurent GS, Wahlestedt C. Noncoding RNAs: couplers of analog and digital information in nervous system function? Trends Neurosci 2007;30:612–21.
[4] Qu Z, Adelson DL. Evolutionary conservation and functional roles of ncRNA. Front Genet 2012;3:205.
[5] Guttman M, Rinn JL. Modular regulatory principles of large noncoding RNAs. Nature 2012;482:339–46.
[6] Volders P-J,Helsens K,Wang X,Menten B,Martens L,Gevaert K, etal.LNCipedia:adatabaseforannotatedhumanlncRNAtranscript sequences and structures. Nucleic Acids Res 2013;41:D246–51.
[7] Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov 2011;1:391–407.
[8] Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet 2009;10:155–9.
[9] Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell 2011;43:904–14.
[10]Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 2010;143:46–58.
[11] Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011;472:120–4.
[12] Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell 2009;136:629–41.
[13] Calin GA, C-g Liu, Ferracin M, Hyslop T, Spizzo R, Sevignani C, et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 2007;12:215–29.
[14] Esteller M. Non-coding RNAs in human disease. Nat Rev Genet 2011;12:861–74.
[15] Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010;464:1071–6.
[16] Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett 2013;339:159–66.
[17] Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Noncoding RNAs: regulators of disease. J Pathol 2010;220:126–39.
[18] Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol 2011;21:354–61.
[19] Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A. A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2004;2:E171.
[20] Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet 2013;9:e1003368.
[21] Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA–protein interactions. Sci China Life Sci 2013;56:876–85.
[22] Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, et al. Largescale prediction of long non-coding RNA functions in a coding–non-coding gene co-expression network. Nucleic Acids Res 2011;39:3864–78.
[23] Khalil AM, Rinn JL. RNA–protein interactions in human health and disease. Semin Cell Dev Biol 2011;22:359–65.
[24] Sacco LD, Baldassarre A, Masotti A. Bioinformatics tools and novel challenges in long non-coding RNAs (lncRNAs) functional analysis. Int J Mol Sci 2011;13:97–114.
[25] Bellucci M, Agostini F, Masin M, Tartaglia GG. Predicting protein associations with long noncoding RNAs. Nat Methods 2011;8:444–5.
[26] Muppirala UK, Honavar VG, Dobbs D. Predicting RNA–protein interactions using only sequence information. BMC Bioinformatics 2011;12:489.
[27] Hearst MA, Dumais ST, Osman E, Platt J, Scholkopf B. Support vector machines. IEEE 1998;13:18–28.
[28] Liaw A, Wiener M. Classification and regression by randomForest. R News 2002;2:18–22.
[29] Lu Q, Ren S, Lu M, Zhang Y, Zhu D, Zhang X, et al. Computational prediction of associations between long noncoding RNAs and proteins. BMC Genomics 2013;14:651.
[30] Suresh V, Liu L, Adjeroh D, Zhou X. RPI-Pred: predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res 2015;43:1370–9.
[31] Ko¨hler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet 2008;82:949–58.
[32] Wang W, Yang S, Zhang X, Li J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics 2014;30:2923–30.
[33] Sarwar B, Karypis G, Konstan J, Riedl J. Item-based collaborative filtering recommendation algorithms. Proceedings of the 10th International Conference on World Wide Web Hong Kong, May 1–5, 2001; ACM 1-58113-348-0/01/0005.
[34] Yuan J, Wu W, Xie C, Zhao G, Zhao Y, Chen R. NPInter v2. 0: an updated database of ncRNA interactions. Nucleic Acids Res 2014;42:D104–8.
[35] Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2. 0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 2013;42:D92–7.
[36] Bhartiya D, Pal K, Ghosh S, Kapoor S, Jalali S, Panwar B, et al. LncRNome: a comprehensive knowledgebase of human long noncoding RNAs. Database 2013;2013:bat034.
[37] Chen X, Yan GY. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics 2013;29:2617–24.
[38] Chen X, Liu MX, Yan GY. Drug–target interaction prediction by random walk on the heterogeneous network. Mol BioSyst 2012;8:1970–8.
[39] Xie C, Yuan J, Li H, Li M, Zhao G, Bu D, et al. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res 2014;42:D98–103.
[40] Zhou T, Ren J, Medo M, Zhang YC. Bipartite network projection and personal recommendation. Phys Rev E: Stat, Nonlin, Soft Matter Phys 2007;76:046115.
*Corresponding author.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer
- EDISON-WMW: Exact Dynamic Programing Solution of the Wilcoxon–Mann–Whitney Test
- Translational Bioinformatics: Past, Present, and Future
- Single-cell Transcriptome Study as Big Data
- Understanding Spatial Genome Organization: Methods and Insights
- Call for Papers Special Issue on‘‘Biomarkers for Human Diseases and Translational Medicine’’