Translational Bioinformatics: Past, Present, and Future
2016-03-31JessicaTenenbaum
Jessica D. Tenenbaum
Translational Bioinformatics: Past, Present, and Future
Jessica D. Tenenbaum*,a
Department of Biostatistics and Bioinformatics, Duke University School of Medicine, Durham, NC 27710, USA
Received 16 December 2015; accepted 20 January 2016
Available online 11 February 2016
Handled by Luonan Chen
E-mail: jessie.tenenbaum@duke.edu (Tenenbaum JD).
aORCID: 0000-0003-3532-565X.
Peer review under responsibility of Beijing Institute of Genomics,
Chinese Academy of Sciences and Genetics Society of China.
http://dx.doi.org/10.1016/j.gpb.2016.01.003
1672-0229ⓒ2016 The Author. Production and hosting by Elsevier B.V. on behalf of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
KEYWORDS
Translational bioinformatics;
Biomarkers;
Genomics;
Precision medicine;
Personalized medicine Abstract Though a relatively young discipline, translational bioinformatics (TBI) has become a key component of biomedical research in the era of precision medicine. Development of high-throughput technologies and electronic health records has caused a paradigm shift in both healthcare and biomedical research. Novel tools and methods are required to convert increasingly voluminous datasets into information and actionable knowledge. This review provides a definition and contextualization of the term TBI, describes the discipline’s brief history and past accomplishments, as well as current foci, and concludes with predictions of future directions in the field.
Introduction
Though a relatively young field, translational bioinformatics has become an important discipline in the era of personalized and precision medicine. Advances in biological methods and technologies have opened up a new realm of possible observations. The invention of the microscope enabled doctors and researchers to make observations at the cellular level. The advent of the X-ray, and later of magnetic resonance and other imaging technologies, enabled visualization of tissues and organs never before possible. Each of these technological advances necessitates a companion advance in the methods and tools used to analyze and interpret the results. With the increasingly common use of technologies like DNA and RNA sequencing, DNA microarrays, and high-throughput proteomics and metabolomics, comes the need for novel methods to turn these new types of data into new information and that new information into new knowledge. That new knowledge, in turn, gives rise to action, providing insights regarding how to treat disease and ideally how to prevent it in the first place.
Translational bioinformatics
Defining translational bioinformatics
According to the American Medical Informatics Association (AMIA), translational bioinformatics (hereafter‘TBI”) is ‘the development of storage, analytic, and interpretive methods to optimize the transformation of increasingly voluminous biomedical data, and genomic data, into proactive, predictive, preventive, and participatory health”(http://www.amia.org/applications-informatics/translational-bioinformatics). Put more simply, it is the development of methods to transform massive amounts of data into health. Dr. Russ Altman from Stanford University delivers a year-in-review talk at AMIA’s summit on TBI. In his 2014 presentation he provided the following definition for TBI:‘informatics methods that link biological entities (genes, proteins, and small molecules) to clinical entities (diseases, symptoms, and drugs)—or vice versa”(https://dl.dropboxusercontent.com/u/2734365/amia-tbi-14-final. pdf). Figure 1 gives a visual depiction of the way in which TBI fits within the bigger picture of biomedical informatics and transforming data into knowledge [1]. Along the X axis is the translational spectrum of bench-to-bedside, while the Y axis from top to bottom represents the central dogma of informatics, transforming data to information and information to knowledge. Toward the discovery end of the spectrum (the bench) is bioinformatics, which includes storage, management, analysis, retrieval, and visualization of biological data, often in model systems. The discovery end of the spectrum has some overlap with computational biology, particularly in the context of systems biology methods. Toward the clinical end of the spectrum (bedside) is health informatics. TBI fits in the middle of this space. On the data-to-knowledge spectrum, data collection and storage are the beginning steps. After that comes data processing, analysis, and then interpretation, thereby transforming the information that has been gleaned from the data into actual knowledge, useful in the context of clinical care, or for further research. In that way the data go from just being ‘bits”—1’s and 0’s—to new knowledge and actionable insights.
Where do we come from? A relatively short past
TBI as a field has a relatively short history. In the year 2000, the initial drafts of the human genome were released, arguably necessitating this new field of study (http://web.ornl.gov/sci/ techresources/Human_Genome/project/clinton1.shtml). In 2002, AMIA held its annual symposium with the name ‘Bio*medical Informatics: One Discipline”, meant to recognize and emphasize the spectrum of subdisciplines. In 2006, the term itself was actually coined by Atul Butte and Rong Chen at the AMIA annual symposium in a paper entitled ‘Finding disease-related genomic experiments within an international repository:first steps in translational bioinformatics”[2]. In 2008, AMIA had its first annual AMIA summit on TBI, chaired by Dr. Butte. Year 2011 saw the first annual TBI Conference in Asia, held in Seoul, Korea. Finally, an online textbook on TBI was published in 2012 by PLoS Computational Biology, edited by Maricel Kann. Initially intended to be a traditional print textbook, this resource was published using an open source model, making it freely available on the Internet (http://collections.plos.org/translational-bioinformatics).

Figure 1 Translational Bioinformatics in contextThe Y axis depicts the‘central dogma”of informatics, converting data to information and information to knowledge. Along the X axis is the translational spectrum from bench to bedside. Translational bioinformatics spans the data to knowledge spectrum, and bridges the gap between bench research and application to human health. The figure was reproduced from [1] with permission from Springer.
What are we? TBI today
A 2014 review article [3] categorized recent themes in the field of TBI into four major categorizations: (1) clinical‘big data”, or the use of electronic health record (EHR) data for discovery (genomic and otherwise); (2) genomics and pharmacogenomics in routine clinical care; (3) omics for drug discovery and repurposing; and (4) personal genomic testing, including a number of ethical, legal, and social issues that arise from such services.
Big data and biomedicine
As technology enables us to take an increasingly comprehensive look across the genome, transcriptome, proteome, etc., the resulting datasets are increasingly high-dimensional. This in turn requires a larger number of samples in order to achieve the statistical power needed to detect the true signal. The past decade or so has seen an increasing number of large-scale biorepositories intended for clinical and translational research all over the world. These projects comprise both information and biospecimens from individual patients, enabling researchers to reclassify diseases based on underlying molecular pathways, instead of the macroscopic symptoms that have been relied on for centuries in defining disease. Examples are listed in Table 1. These various projects involve different models of participation, ranging from explicit informed consent to use of de-identified biospecimens and their associated clinical information from EHRs (also de-identified). The informed consent is the most ethically rigorous model, but also the most expensive. The use of de-identified specimens and data is more scalable, and financially feasible. However, as complete genomic data are increasingly used, it is impossible to truly deidentify these data [4]. This raises ethical issues regarding patient privacy and data sharing. In the United States, legislation known as the‘Common Rule”addresses these issues. In 2015, a notice of proposed rule-making (NPRM, http://www. hhs.gov/ohrp/humansubjects/regulations/nprmhome.html) was released to solicit feedback on some major revisions to the law, which was originally passed in 1991. Much has changed within biomedical research in the intervening years [5].
In order to accrue the numbers of samples required for the ‘big data”discipline that biomedical research is becoming, the ability to use patient data and samples in research would be of significant benefit. One major point addressed in theaforementioned NPRM is the ability for patients to give broad consent for future use of data and samples, without knowing the specifics of research studies ahead of time. As we move toward the learning healthcare system (LHS) model [6] in which every encounter is an additional data point, explicit research registries will become less relevant. They will be too expensive to maintain, and larger numbers of patients/participants will be available through federated initiatives that allow a researcher to query across institutions regionally, nationally, and even internationally. The National Patient-Centered Clinical Research Network (PCORnet) takes this approach, enabling clinical outcome research through federated pragmatic clinical trials. Importantly, this initiative emphasizes partnership with patients and their advocates, so that they are empowered as collaborators, with a say in what research questions matter most [7].

Table 1 Large-scale research initiatives integrating human specimens with clinical annotation
LHS is about moving from evidence-based practice, i.e., clinical care decisions based on conscientious use of current best evidence, to practice-based evidence, i.e., the generation of evidence through collection of data in the real-world as opposed to the artificially-controlled environment of randomized clinical trials [8]. In recent decades, the biomedical enterprise has strived to practice medicine in a way that is supported by the best possible evidence from randomized clinical trials. But clinical trials have their own issues. They are expensive, and they tend to be very different from real life scenarios [9]. Criteria for inclusion in a trial often include the absence of common comorbidities or use of common medications. Compliance tends to be high, but the cohort being studied is often not representative of the target population for the treatment in question. In LHS, translation becomes bi-directional. Research is used to inform practice, whereas data that are generated in the course of clinical care can in turn be used for both hypothesis generation and validation through pragmatic trials. Data derived from clinical care can thus inform clinical guidelines and future practice.
Secondary use Secondary use of data refers to data that are created or collected through clinical care. In addition to use in caring for the patient, these data may also be crucial for operations, quality improvement, and comparative effectiveness research. Some assert that the term‘secondary use”should give way to the term‘continuous use.”They argue against the notion that data collected at the point of care are solely for clinical use, and everything else is secondary. We should be maximally leveraging this valuable information. Nonetheless, there is a legitimate concern about data quality. Data in the EHR are often sparse, incomplete, even inaccurate [10]. This makes these data wholly unsuitable for certain purposes, but still sufficient for others. For instance, Frankovich et al. described a case in which an adolescent lupus patient was admitted with a number of complicating factors that put her at risk for thrombosis [11]. The medical team considered anticoagulation, but were concerned about the patient’s risk of bleeding. No guideline was available for this specific case, and a survey of colleagues was inconclusive. Through the institution’s electronic medical record data warehouse, Frankovich and colleagues were able to look at an‘electronic cohort”of pediatric lupus patients who had been seen over a 5-year period. Of the 98 patients in the cohort, 10 patients had developed clots, with higher prevalence in patients with similar complications as the patient in question. Using this real-time analysis based on evidence generated in the course of clinical care, Frankovich and colleagues were able to make an evidencebased decision to administer anti-coagulants [11]. Subsequently, researchers at Stanford University have proposed a ‘Green Button”approach to formalize this model of realtime decision support derived from aggregate patient data and data capture to help inform future research and clinical decisions [12].
TBI tends to focus on molecules, newly accessible in high dimensions based on novel high-throughput technologies. Phenotyping is a closely-related challenge, more complex than it might seem. Disease is not binary: even within a very specific type of cancer, a tumor’s genomic profile may be quite different among the precise sampling locations and sizes [13]. There are a number of groups focusing on this problem: the Electronic Medical Records and Genomics (eMERGE) Network (http://www.genome.gov/27540473), the NIH Collaboratory (https://www.nihcollaboratory.org/), PCORnet (http://www.pcornet.org/), and the MURDOCK Study (http://murdock-study.com/), among others [14–17]. The Phenotype KnowledgeBase website (https://phekb.org/) is a knowledge base of phenotypes, offering a collaborative environment to build and validate phenotype definitions. The phenotypes are not (yet) computable, but it serves as a resource for defining patient cohorts in specific disease areas [18]. Richesson et al. looked at type 2 diabetes, a phenotype that one might expect to be fairly straightforward [19]. But defining type 2 diabetes mellitus (T2DM) using the International Classification of Disease version 9 (ICD9) codes, diabetes-related medications, the presence of abnormal labs, or a combination of those factors resulted in very different counts for the number of people diagnosed with T2DM in Duke’s data warehouse [19]. Using only ICD9 codes gave 18,980 patients, while using medications yielded 11,800. Using ICD9 codes, medications, and labs all together yielded 9441 patients. Note that the issue is not just a matter of semantics and terminology, where if everyone could agree to a single definition and use the same code, then the terms would become uniform. For different purposes, different definitions of diabetes may be needed, depending on whether the use case involves cohort identification or retrospective analysis. In different cases, one might care more about minimizing false positives (e.g., retrospective analysis) or maximizing true positives (e.g., surveillance or prospective recruitment).
Thousands of papers have been published describing genome-wide association studies (GWAS), in which researchers look across the entire genome to find SNPs that are statistically enriched for a given phenotype (usually a disease) compared with healthy controls [20]. Researchers at Vanderbilt University turned this approach on its head, developing a method known as phenome-wide association studies (PheWAS, https://phewas.mc.vanderbilt.edu/). Instead of looking at the entire genome, PheWAS evaluates the association between a set of genetic variants and a wide and diverse range of phenotypes, diagnoses, traits, and/or outcomes [21]. This analytic approach asks, for a given variant, do we see an enrichment of a specific genotype in any of these phenotypes? Figure 2 illustrates results using this approach [22]. In standard GWAS analyses, the different color bands at the bottom represent the different chromosomes. In the case of PheWAS, they are different disease areas, e.g., neurologic, cardiovascular, digestive, and skin. Pendergrass et al. [23] used a PheWAS approach for the detection of pleiotropic effects, where one gene affects multiple different phenotypes. They were able to replicate 52 known associations and 26 closely-related ones. They also found 33 potentially-novel genotype-phenotype associations with pleiotropic effects, for example the GALNT2 SNP that had previously been associated with HDL levels among European Americans. Here they detected an association between GALNT2 and hypertension phenotypes in African Americans, as well as serum calcium levels and coronary heart disease phenotypes in European Americans.
Another aspect of big data in biomedicine is the use of nontraditional data sources. These were well illustrated, both literally and figuratively, in a 2012 paper by Eric Schadt [24]. A complex and detailed figure (Figure 3) showed various data types that could be mined for their effects on human health: weather, air traffic, security, cell phones, and social media among others. But strikingly to those reading the paper just a few years later, the list did not include personal activity trackers, e.g., FitBit, Jawbone, or even the Apple watch. This omission of such a popular technology today is indicative of what a fast-moving field this is.
Genomics in clinical care
One sees a number of examples of how genomic data are used in clinical care in the context of pharmacogenomics [25]. But molecular data, and genomic data derived from nextgeneration sequencing (NGS) in particular, have been used in a number of other contexts as well. One example took place at Stanford’s Lucile Packard Children’s Hospital, where a newborn presented with a condition known as long QT syndrome. (http://scopeblog.stanford.edu/2014/06/30/whenten-days-a-lifetime-rapid-whole-genome-sequencing-helps-criti-cally-ill-newborn/). In this specific case, the manifestation was unusually severe—the baby’s heart stopped multiple times in the hours after its birth. Long QT syndrome can be caused by mutations in a number of different genes [26]. It is necessary to know which gene harbors the mutation in order to know how to treat the condition [26]. In this case, a whole-genome sequencing (WGS) was performed enabling identification of a previously-studied mutation, as well as a novel copy number variation in the TTN gene that would not otherwise have been detectable through targeted genotyping alone. Moreover, NGS enabled the answer to be obtained in a matter of hours to days instead of weeks.

Figure 2 A PheWAS Manhattan plot for a given SNPThis plot shows the significance of association between SNP rs965513 and 866 different phenotypes. Along the X axis different disease groups are shown in different colors. This is in contrast to an analogous plot for GWAS in which the X axis would represent the different chromosomes. The Y axis reflects the P value for each phenotype. Blue and red horizontal lines represent P value of 0.05 and Bonferroni corrected P value of 5.8×10-5, respectively. PheWAS, phenome-wide association studies; GWAS, genome-wide association studies. The figure was reproduced from [22] with permission from Elsevier.
Another example of DNA sequencing in clinical care involved diagnosis of infectious disease. A 14-year-old boy with severe combined immunodeficiency (SCID) had been admitted to the hospital repeatedly. He had headache, fever, weakness, nausea, and vomiting. His condition continued to decline to the point where he was put into a medicallyinduced coma. A normal diagnostic work up was‘unrevealing”and the doctors were unable to determine the etiology of his condition. The patient was enrolled in a research study for pathogen detection and discovery in hospitalized patients. The protocol for this study involved performing NGS on the subject’s spinal fluid. The results included detection of 475 reads corresponding to leptospira infection. Of note, the normal test for leptospirosis involves detection of the patient’s antibody response to the infection. In this case, the patient’s SCID status prevents such a response, so the infection was not detectable through standard means [27].
Cancer has been one of the most active areas for the translation of genomic discoveries into changes in clinical care. One of the biggest players in this space is Foundation Medicine, which makes the FoundationOne test, a targeted panel that uses NGS to test all genes known to harbor mutations in solid tumors. Johnson et al. [28] looked retrospectively at approximately 100 patients who had undergone FoundationOne, and found that 83% of them had potentially actionable results from that test, of which 21% received genotype-directed treatment. Explanations for why the indicated treatment was not given in 79% of those cases included the decision to use standard therapy and clinical deterioration. These results indicate that application of genomic technology has transcended the research domain. In many cases, the findings are clinically actionable. Mirroring this fact, it is worth noting that somemedical insurance companies in the United States started to offer coverage of the FoundationOne test in late 2014 (http://www.clinicalinformaticsnews.com/2014/10/17/foundationmedicine-wins-insurance-coverage-priority-health.html).
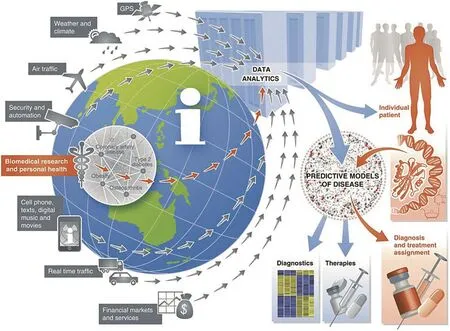
Figure 3 Heterogeneous and non-traditional sources of big dataTechnological advances have enabled the collection and storage of big data beyond biomedicine, including everything from credit card transactions to security cameras to weather. Notably absent from this 2012 figure are smart watches and fitness tracking devices, which became pervasive in the years that followed. The figure was reproduced from [24] under Creative Commons Attribution license.
In addition to targeted panels, deep cancer sequencing can help shed light on drug sensitivity and resistance. Wagle et al. [29] describe a case of a 57-year-old woman with anaplastic thyroid cancer. Her specific tumor was initially sensitive to everolimus, a mammalian target of rapamycin (mTOR) inhibitor. Her doctors were able to sequence the tumor before it became resistant, revealing a mutation in TSE2, which encodes a negative regulator of mTOR. Normally mTOR is downregulated by TSE2, but the mutation caused TSE2 not to down-regulate mTOR to a sufficient level. Therefore, everolimus, which inhibits mTOR, was effective in treating this specific cancer. Later this patient’s cancer became drug-resistant, whereupon the newly drug-resistant part of the tumor was sequenced again. It was discovered that an mTOR mutation had cause mTOR not to be inhibited by allosteric inhibitors like everolimus. An allosteric inhibitor binds to the protein in question somewhere other than on the active site of the molecule. Knowing the specific cause for the newly-acquired resistance leads to other treatment options. They were able to switch to mTOR kinase inhibitors, in order to downregulate the pathway through other mechanisms.
Despite these compelling cases, it is worth noting that at this point, tumor sequencing is the exception in clinical oncology, and not one of the routine procedures. Only after‘first line”treatment has failed are tumors sequenced, and even that is largely confined to large academic medical centers. It will be interesting to see if, when, and how that changes.
While blood clotting and cancer are the areas where most actionable pharmacogenomics findings have been made, a notable exception is work by Tang and his colleagues [30], in which they describe the identification of a genotype-based treatment for T2DM with an alpha2A-adrenergic receptor antagonist. A specific genetic variant causes over-expression of the alpha2A-adrenergic receptor, and impaired insulin secretion. They hypothesized that if they could block the overexpressed receptor, they could increase insulin secretion. The authors looked at patients with and without the mutation in question. Those with the mutation showed a dose response to the drug as measured through levels of insulin. Participants without the mutation showed no such response. This research is especially interesting because it goes beyond cancer and blood thinners, into a chronic disease that is affecting an increasing portion of the world’s population.
One of the earliest and best known DNA sequencing success stories was that of Nic Volker, who at age 2 developed severe gastro-intestinal issues resembling Crohn’s disease, for which a diagnosis could not be determined. His story was covered in the Milwaukee Journal Sentinel in 2011 in a piece that was later awarded a Pulitzer Prize for explanatory reporting (http://www.jsonline.com/news/health/111641209.html). After multiple life-threatening situations and a protracted diagnostic odyssey, Volker’s genome was sequenced to look for a causal mutation. Doing so enabled the discovery of a mutation in the gene X-linked inhibitor of apoptosis (XIAP). Equally important, there existed a known therapeutic intervention for disorders caused by XIAP mutations. A progenitor cell transplant was performed and the patient’s condition improved. Though Nic is not without ongoing health challenges, he celebrated his 11th birthday in October, 2015.
Despite the successes described above, there is also reason for caution regarding use of NGS sequencing in clinical care. Dewey et al. [31] performed Illumina-based WGS on 12 different participants. Confirmatory sequencing was formed on 9 of those participants by Complete Genomics Inc. Their findingsincluded the fact that 10%–19% of inherited disease genes were not covered to accepted standards for SNP discovery. For the genotypes that were called, concordance between the two technologies for previously-described single nucleotide variants was 99%–100%. However, for insertion and deletion variants, the concordance rate was only 53%–59%. Approximately 15% were discordant, and approximately 30% of the variants could not be called by one technology or the other. In addition, inter-rater agreement for whether findings should be followed up clinically was only 0.24 (‘fair”) by Fleiss’kappa metric. Rater agreement was even worse for cardiometabolic diseases, with the rate at which the two experts agreed on the need for clinical follow-up worse than random in those cases. Notably, the estimated median cost for sequencing and variant interpretation was about US $15,000, plus the price of the computing infrastructure and data storage. This means the cost of interpretation is significantly more than the proverbial US $1000 genome goal, but is also significantly less than the US $100,000 or $1 million some had feared [32].
Omics for drug discovery and re-purposing
Much has been said about the protracted process involved in getting a drug through the FDA approval pipeline. Estimates are that the process can take on average 12 years between lead identification and FDA approval. This makes the prospect of drug repurposing an appealing one. Drug repurposing refers to taking an existing, FDA-approved compound and using it to treat a disease or condition other than the one for which it was originally intended [33]. In the past, inspiration for this type of‘off label use”has been largely serendipitous. For example, Viagra was initially aimed at treating heart disease, and turned out to be useful for erectile dysfunction [34]. By using a pre-approved compound, early phase clinical trials can be avoided, which can save significant time and money.
Computational approaches to drug repurposing may take a number of different forms as described in two recent reviews [33,35]. One is to look for molecular signatures in disease and compare those to signatures observed in cells, animal models, or people who have been treated with different drugs. If anti-correlated signatures can be identified between diseases and drugs, administration of that drug for that disease may help cure the condition, or at least to alleviate the symptoms. One of the prominent early examples of a computational approach to drug repositioning was the Broad Institute’s Connectivity Map (CMap) [36]. The authors identified gene expression signatures for disease states and perturbation by small molecules and then compared those signatures. They made the data available as a resource intended to enable the identification of functional connections between drugs, genes, and diseases. Another example is work by Jahchan et al. [37], in which they identified anti-depressants as potential inhibitors of lung cancer. The authors looked at numerous disease and drug profiles and found an anti-correlation between gene expression seen after administration of anti-depressants and the pattern of expression observed in small cell lung cancer. They next transplanted these types of tumors into mice and found that with the drug, those tumors either shrunk or didn’t even grow. They also used indigenous tumors in the mouse model for this type of cancer and found that the anti-depressant had promising results on those tumors and for other endocrine tumors as well. They were able to start a clinical trial, much faster than would have been possible by chance or by various other traditional methods, though unfortunately that trial was ultimately terminated for lack of efficacy. A similar approach was used by Sirota et al. to identify the anti-ulcer drug cimetidine as a candidate agent to treat lung adenocarcinoma and validate this off-label usage in vivo using an animal model of the lung cancer [38].
An alternative to the largely computational methods described above is an experimental approach to drug repositioning. For example, Nygren et al. screened 1600 known compounds against 2 different colon cancer cell lines [39]. They used Connectivity Map data to further evaluate their findings, and identified mebendazole (MBZ) as having potential therapeutic effect in colon cancer. Finally, Zhu et al. mined data from PharmGKB [40] and leveraged the web ontology language (OWL) to perform semantic inference. They were able to identify potential novel uses and adverse effects of approved breast cancer drugs [41].
Personalized genomic testing
The year 2008 saw the founding of several companies that offered direct-to-consumer (DTC) genetic testing, reporting on a variety of genes for both health and recreational purposes. As of 2016, 23andMe was the last major player standing in the United States, with other companies having been acquired and/or changing their business models.
DTC genetic testing raises a number of interesting ethical, legal, and social issues. For several years, there was an open question as to whether or not these tests should be subject to government regulation. In November 2013, the US FDA ordered 23andMe to stop advertising and offering their health-related information services. The FDA considered these tests to be‘medical devices”and as such to require formal testing and FDA approval for each test. In February 2015, it was announced that the FDA had approved 23andMe’s application for a test for Bloom syndrome (http://www.fda.gov/News Events/Newsroom/PressAnnouncements/UCM435003), and in October2015itwasannouncedthatthecompanywouldonce againbeofferinghealthinformationintheformofcarrierstatus for36genes[42].Notethata23andMecustomerisabletodownload his or her raw genomic data and to use information from other websites to interpret the results, including Promethease (http://www.snpedia.com/index.php/Promethease), Geneticgenie (http://geneticgenie.org/), openSNP (https://opensnp.org/), and Interpretome (http://interpretome.com/) for healthrelated associations.
Another important question raised by DTC genetic testing include whether the consumers are ready for this information. Traditionally, patients receive troubling health-related information in a face-to-face conversation with their doctor. There is some concern that patients are not competent or wellequipped to receive potentially distressing news through an Internet link [43]. To help mitigate this concern, 23andMe ‘locks”certain results, making them accessible only if the user clicks through an additional link, indicating they truly want to know.
What about the healthcare providers? Are they ready to incorporate genomic data, patient-supplied or otherwise,into treatment decisions? In a case described in 2012, a 35-year-old woman informed her fertility care provider that her 23andMe results revealed a relatively common (1 in 100) blood clotting mutation [44]. She was surprised when her provider responded by saying that, were she to become pregnant, she would need to be put on an injectable anti-coagulant throughout her pregnancy. With no family history of blood clotting disorders, nor personal history of recurrent miscarriage, this mutation would have gone untested, had it not been for the DTC results. However, when this patient did become pregnant and consulted with a specialist whose expertise was in blood clotting disorders in pregnant women, the anticoagulant was indeed prescribed. Note that the guidelines have since changed, and the prophylactic treatment for clotting would not be prescribed today. It is also worth noting that in the United States, the Genetic Information Nondiscrimination Act (GINA) prevents employers and health insurers from discriminating against anyone based on their genetic information. It does not, however, cover long-term care, disability, or life insurance. Therefore when this woman applied for life insurance after her twins were born, the rate she was offered was more than twice what it would have been had she not known about the blood clotting mutation and been treated prophylactically for the increased risk it conferred [Tenenbaum and colleagues, unpublished data].
A more positive example of where genetic testing is helping patients is a case presented at the American Neurological Association conference in 2014. A patient had a history of Alzheimer’s disease on her mother’s side of the family. She did not know if she was a carrier, nor did she want to know. But she wanted to ensure that she did not pass that mutation to her future children. Preimplantation genetic diagnosis (PGD) testing enabled her doctors to select embryos that did not have that Alzheimer’s disease gene mutation. The patient herself was never tested, nor was she informed how many (if any) of the embryos contained the mutation. (http://www. wsj.com/articles/genetic-testing-for-alzheimerswithout-revealingthe-results-1413221509).
Privacy
A 2013 Science paper from the Erlich lab at Massachusetts Institute of Technology (MIT) generated much controversy when the authors demonstrated the ability to re-identify a number of individuals using publicly-available genealogy databases and genetic data [45]. The researchers used the short tandem repeat (STR) data from the Y chromosome, year of birth, and state of residence, combined with information from public genealogy websites to identify individuals. They did this by starting with publicly-available STRs and entering them on genealogy websites to identify matches. Note that their accuracy was not 100%. Though they were able to identify Craig Venter based on his genomic data, they failed to identify several other individuals, particularly those with more common last names. Overall, they reported a 12% success rate in recovering surnames of US males. They were also able to reconstruct Utah family pedigrees based on 1000 Genomes Project data and other publicly-available sources. Due to a number of cultural and historical factors, families in Utah tend to be large and genealogically well-documented, and an unusually high proportion of the Utah population has participated in scientific studies involving genomic data. It is worth noting that the researchers did not release any names that were not previously public, nor did they use the information for any nefarious purposes. Their primary interest was to demonstrate that such re-identification was possible. Interestingly, and somewhat surprisingly, the terms of use for the datasets did not prohibit re-identification.
Where are we going? The road ahead
Though we cannot know what the future holds, we can make some informed guesses based on events to date. The author believes that in the not-too-distant future, newborns will be sequenced at birth, just as we currently test for a more limited number of genetic issues. With the cost of sequencing a genome still at or above US $1000, such widespread sequencing is not yet realistic. But researchers are already performing pilot studies in this area, to better understand and anticipate the issues that are likely to arise. As an example, the MedSeq project at Harvard University is a study designed both to integrate WGS into clinical care and to assess the impact of doing so [46]. In addition, the Geisinger Health System has partnered with Regeneron on a project known as MyCode, which aims to sequence the exomes of 250,000 patients in the Geisinger system. In late 2015, the project began returning results to patients for 76 genes (https://www.genomeweb.com/sequencing-technology/geisinger-begins-returning-clinically-actionableexome-sequencing-results).
Even clearer is that tumor sequencing will be performed as part of standard of care for cancer. Currently sequencing is performed at certain tertiary and quaternary care facilities, particularly for metastatic tumors. As more is discovered about the various dysregulated pathways in cancer, and about the therapeutic implications for different genetic variations, the blunt mallet that is chemotherapy will be phased out in favor of far more precise and targeted therapies.
The microbiome has seen increasing attention in recent years, a trend that will certainly continue for the foreseeable future. It is not surprising that the make-up of the microbial communities that likely outnumber the cells of the human body [47] can have significant impact on human health, particularly in metabolic and gastro-intestinal disease. The more surprising trend is the connection between the microbiome and other, more unexpected phenotypes, for example, anxiety, depression, and autism [48,49]. This is likely to continue as this area of research continues to grow.
We will continue to see an increase in analyses of different ‘omic”types. Genomics has been by far the most popular area of focus to date. As technologies mature, we will continue to see biomarker discovery in proteomics, metabolomics, and other as-yet-unnamed‘omic”modalities. We will also see increasingly integrated analysis, taking a systems approach to human biology where to date systems biology has been focused on model organisms, often single cellular ones, in which the system can be methodologically, and ethically, perturbed. Early examples of integrative, multi-modal analysis include the integration of microRNAs and transcription factors to determine regulatory networks underlying coronary artery disease [50], integrative analysis of genomics and transcriptomics to look at cardiovascular disease [51], and theuse of metabolomics data with GWAS to elucidate molecular pathways [52].
The coming decade will see more biomarker-based research and insights into mental health disorders. To date, cancer and cardiology have received significant attention, to great advantage. But those disease areas are, by comparison, relatively easy to identify, to distinguish, and even to quantify. This is not the case for neurological and psychiatric disorders. Mental health is an area where diagnosis, and phenotyping more generally, is as much art as science. It is an area that poses enormous burdens on society, both financial and quality-of-life related, and is also ripe for a deeper, more physiologically-based understanding [53]. Even if therapy is still a long way away, having some concrete, quantifiable biomarkers, by which we could classify conditions such as depression, bi-polar disorder, and manic-depressive tendencies, would be a great leap.
Finally, major changes will be required to effectively and efficiently train the workforce of tomorrow. These changes will not simply entail adding a few quantitative courses into medical and graduate level biomedical research training, though that too will be important. The need for more informatics professionals is being addressed to some extent by the significant number of training programs being created at multiple different levels, from certificates to master’s to PhD programs. In addition to the informatics workforce, more genetic counselors are needed. It is estimated that there are only approximately 3500 genetic counselors in the entire United States (http:// www.abgc.net/About_ABGC/GeneticCounselors.asp). As genomic sequencing becomes increasingly widespread, this number needs to increase both in the US and around the globe. Genetic counselors today may be compared to pathologists in the early days of the microscope, or radiologists in the early days of X-rays. The respective numbers will never need to be equal—except in the case of cancer, the genome need only be sequenced once in a person’s lifetime. Moreover, pathologists and radiologists detect and describe what is. Even as we learn more about our genomes, they primarily tell us what is more or less likely to be. Still, many genetic findings are already actionable today, and this number will continue to increase.
There are two areas where we need to do better, but I am less optimistic that we will see real progress in the next decade or so. The first is in adoption of data standards. We need better resources for understanding, navigating, and using existing standards [54]. We also need more impactful incentives for adoption, and disincentives for failure to do so. In the current landscape, standards are too difficult to identify and adopt, and the benefit of doing so tends to be realized by people other than those doing the hard work.
Lastly, we need to establish more inter-interdisciplinary coordination and collaboration. Perhaps meta-interdisciplinary isabetterterm.Asbiomedicalinformaticians,wearebydefinition interdisciplinary, including training and perspectives from medicine,biology,andcomputerscience.Buttherearealargenumber of different communities around the world who are working on these problems, talking mostly among themselves. The various professional societies, even the interdisciplinary ones, have their respective meetings. There is some cross pollination, and some overlap in who attends the respective events but still not enough. There could be so much more, and we could make significant progress, reduce redundancy, and increase return on investment for research funding if these groups could be more consciously and proactively in sync.
Conclusion
In summary, we are entering a new era in data-driven health care. Translational bioinformatics methods continue to make an actual difference in patients’lives. The infrastructure, information technology, policy, and culture need to catch up with some of the technological advances. For researchers working at the cutting edge of translational bioinformatics, opportunities abound, and the future looks bright.
Competing interests
The author has declared no competing interests.
Acknowledgments
This work was supported in part by the Clinical and Translational Science Award (Grant No. UL1TR001117) to Duke University from the National Institutes of Health (NIH), United States.
References
[1] Tenenbaum JD, Shah NH, Altman RB. Translational bioinformatics. In: Shortliffe EH, Cimino JJ, editors. Biomedical informatics. London: Springer-Verlag; 2014. p. 721–54.
[2] Butte AJ, Chen R. Finding disease-related genomic experiments within an international repository:first steps in translational bioinformatics. In: AMIA Annual Symposium Proceedings 2006. p. 106–10.
[3] Denny JC. Surveying recent themes in translational bioinformatics: big data in EHRs, omics for drugs, and personal genomics. Yearb Med Inform 2014;9:199–205.
[4] Kulynych J, Greely H. Every patient a subject: When personalized medicine, genomic research, and privacy collide, 2014, http:// www.slate.com/articles/technology/future_tense/2014/12/when_ personalized_medicine_genomic_research_and_privacy_collide.
[5] Hudson KL, Collins FS. Bringing the common rule into the 21st century. N Engl J Med 2015;373:2293–6.
[6] Institute of Medicine (US) Roundtable on Evidence-Based Medicine. Leadership commitments to improve value in healthcare:finding common ground: workshop summary. Washington (DC): National Academies Press (US); 2009, http://www.ncbi. nlm.nih.gov/books/NBK52847/.
[7] Fleurence R, Selby JV, Odom-Walker K, Hunt G, Meltzer D, Slutsky JR, et al. How the patient-centered outcomes research institute is engaging patients and others in shaping its research agenda. Health Aff 2013;32:393–400.
[8] Embi PJ, Payne PR. Evidence generating medicine: redefining the research-practice relationship to complete the evidence cycle. Med Care 2013;51:S87–91.
[9] Luce BR, Kramer JM, Goodman SN, Connor JT, Tunis S, Whicher D, et al. Rethinking randomized clinical trials for comparative effectiveness research: the need for transformational change. Ann Intern Med 2009;151:206–9.
[10] Botsis T, Hartvigsen G, Chen F, Weng C. Secondary use of EHR: data quality issues and informatics opportunities. AMIA Jt Summits Transl Sci Proc 2010;2010:1–5.
[11] Frankovich J, Longhurst CA, Sutherland SM. Evidence-based medicine in the EMR era. N Engl J Med 2011;365:1758–9.
[12] Longhurst CA, Harrington RA, Shah NH. A‘green button’for using aggregate patient data at the point of care. Health Aff 2014;33:1229–35.
[13] Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92.
[14] Kho AN, Pacheco JA, Peissig PL, Rasmussen L, Newton KM, Weston N, et al. Electronic medical records for genetic research: results of the eMERGE consortium. Sci Transl Med 2011;3:79re1.
[15] Richesson RL, Hammond WE, Nahm M, Wixted D, Simon GE, Robinson JG, et al. Electronic health records based phenotyping in next-generation clinical trials: a perspective from the NIH Health Care Systems Collaboratory. J Am Med Inform Assoc 2013;20:e226–31.
[16] Collins FS, Hudson KL, Briggs JP, Lauer MS. PCORnet: turning a dream into reality. J Am Med Inform Assoc 2014;21:576–7.
[17] Tenenbaum JD, Christian V, Cornish MA, Dolor RJ, Dunham AA, Ginsburg GS, et al. The MURDOCK study: a long-term initiative for disease reclassification through advanced biomarker discovery and integration with electronic health records. Am J Transl Res 2012;4:291–301.
[18] Rasmussen LV, Thompson WK, Pacheco JA, Kho AN, Carrell DS, Pathak J, et al. Design patterns for the development of electronic health record-driven phenotype extraction algorithms. J Biomed Inform 2014;51:280–6.
[19] Richesson RL, Rusincovitch SA, Wixted D, Batch BC, Feinglos MN, Miranda ML, et al. A comparison of phenotype definitions for diabetes mellitus. J Am Med Inform Assoc 2013;20: e319–26.
[20] Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 2009;106:9362–7.
[21] Pendergrass SA, Ritchie MD. Phenome-wide association studies: leveraging comprehensive phenotypic and genotypic data for discovery. Curr Genet Med Rep 2015;3:92–100.
[22] Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet 2011;89:529–42.
[23] Pendergrass SA, Brown-Gentry K, Dudek S, Frase A, Torstenson ES, Goodloe R, et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture using Genomics and Epidemiology (PAGE) network. PLoS Genet 2013;9:e1003087.
[24] Schadt EE. The changing privacy landscape in the era of big data. Mol Syst Biol 2012;8:612.
[25] McCarthy JJ, McLeod HL, Ginsburg GS. Genomic medicine: a decade of successes, challenges, and opportunities. Sci Transl Med 2013;5:189sr4.
[26] Baskar S, Aziz PF. Genotype-phenotype correlation in long QT syndrome. Glob Cardiol Sci Pract 2015;2015:26.
[27] Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, Yu G, et al. Actionable diagnosis of neuroleptospirosis by nextgeneration sequencing. N Engl J Med 2014;370:2408–17.
[28] Johnson DB, Dahlman KH, Knol J, Gilbert J, Puzanov I, Means-Powell J, et al. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist 2014;19:616–22.
[29] Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med 2014;371:1426–33.
[30] Tang Y, Axelsson AS, Spegel P, Andersson LE, Mulder H, Groop LC, et al. Genotype-based treatment of type 2 diabetes with an alpha2A-adrenergic receptor antagonist. Sci Transl Med 2014;6:257ra139.
[31] Dewey FE, Grove ME, Pan C, Goldstein BA, Bernstein JA, Chaib H, et al. Clinical interpretation and implications of whole-genome sequencing. JAMA 2014;311:1035–45.
[32] Mardis ER. The $1,000 genome, the $100,000 analysis? Genome Med 2010;2:84.
[33] Shameer K, Readhead B, Dudley JT. Computational and experimental advances in drug repositioning for accelerated therapeutic stratification. Curr Top Med Chem 2015;15:5–20.
[34] Ban TA. The role of serendipity in drug discovery. Dialogues Clin Neurosci 2006;8:335–44.
[35] Li J, Zheng S, Chen B, Butte AJ, Swamidass SJ, Lu Z. A survey of current trends in computational drug repositioning. Brief Bioinform 2015;17:2–12.
[36] Lamb J. The connectivity map: a new tool for biomedical research. Nat Rev Cancer 2007;7:54–60.
[37] Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov 2013;3:1364–77.
[38] Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, et al. Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med 2011;3:96ra77.
[39] Nygren P, Fryknas M, Agerup B, Larsson R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J Cancer Res Clin Oncol 2013;139:2133–40.
[40] Altman RB. PharmGKB: a logical home for knowledge relating genotype to drug response phenotype. Nat Genet 2007;39:426.
[41] Zhu Q, Tao C, Shen F, Chute CG. Exploring the pharmacogenomics knowledge base (PharmGKB) for repositioning breast cancer drugs by leveraging web ontology language (OWL) and cheminformatics approaches. Pac Symp Biocomput 2014:172–82.
[42] Hayden EC. Out of regulatory limbo, 23andMe resumes some health tests and hopes to offer more. Nature 2015. http://dx.doi. org/10.1038/nature.2015.18641.
[43] Green RC, Roberts JS, Cupples LA, Relkin NR, Whitehouse PJ, Brown T, et al. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med 2009;361:245–54.
[44] Tenenbaum JD, James A, Paulyson-Nunez K. An altered treatment plan based on Direct to Consumer (DTC) genetic testing: personalized medicine from the patient/pin-cushion perspective. J Pers Med 2012;2:192–200.
[45] Gymrek M, McGuire AL, Golan D, Halperin E, Erlich Y. Identifying personal genomes by surname inference. Science 2013;339:321–4.
[46] Vassy JL, Lautenbach DM, McLaughlin HM, Kong SW, Christensen KD, Krier J, et al. The MedSeq project: a randomized trial of integrating whole genome sequencing into clinical medicine. Trials 2014;15:85.
[47] Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell 2016;164:337–40.
[48] Foster JA, Neufeld K-AM. Gut–brain axis: how the microbiome influences anxiety and depression. Trends Neurosci 2013;36:305–12.
[49] Mulle JG, Sharp WG, Cubells JF. The gut microbiome: a new frontier in autism research. Curr Psychiatry Rep 2013;15:1–9.
[50] Zhang Y, Liu D, Wang L, Wang S, Yu X, Dai E, et al. Integrated systems approach identifies risk regulatory pathways and key regulators in coronary artery disease. J Mol Med 2015;93:1381–90.
[51] Yao C, Chen BH, Joehanes R, Otlu B, Zhang X, Liu C, et al. Integromic analysis of genetic variation and gene expressionidentifies networks for cardiovascular disease phenotypes. Circulation 2015;131:536–49.
[52] Suhre K, Raffler J, Kastenmuller G. Biochemical insights from population studies with genetics and metabolomics. Arch Biochem Biophys 2015;589:167–76.
[53] Insel TR, Cuthbert BN. Medicine. Brain disorders? Precisely. Science 2015;348:499–500.
[54] Tenenbaum JD, Sansone SA, Haendel M. A sea of standards for omics data: sink or swim? J Am Med Inform Assoc 2014;21:200–3.
REVIEW
*Corresponding author.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer
- Understanding Spatial Genome Organization: Methods and Insights
- Single-cell Transcriptome Study as Big Data
- A Bipartite Network-based Method for Prediction of Long Non-coding RNA–protein Interactions
- EDISON-WMW: Exact Dynamic Programing Solution of the Wilcoxon–Mann–Whitney Test
- Call for Papers Special Issue on‘‘Biomarkers for Human Diseases and Translational Medicine’’