并指(趾)畸形的分类及遗传学研究进展
2016-03-30刘金秀陈玮王香荣周清司彪段文元
刘金秀,陈玮,王香荣,周清,司彪,段文元
并指(趾)畸形的分类及遗传学研究进展
刘金秀,陈玮,王香荣,周清,司彪,段文元
【摘要】并指(趾)畸形(syndactyly,SD)是最常见的遗传性肢体畸形,表现为某些手指或脚趾的融合。这类畸形可单独出现,也可以作为300多种综合征的一个体征出现。SD在家庭间及家庭内部具有显著的临床异质性。即使在同一个体中,表型也可表现为单侧或双侧,对称或不对称。目前,已经报道的非综合征型的SD至少9种,主要遗传模式为常染色体显性遗传,其次是常染色体隐性遗传及X-连锁隐性遗传。其中Ⅱ-a,Ⅲ,Ⅳ,Ⅴ,Ⅶ,Ⅷ型的SD致病基因及其突变位点已经找到,其他SD的遗传学机制仍然未知。在Sajid Malik分类的基础上,系统总结近几年SD的遗传研究成果,以期为该病机制的深入研究和临床诊断提供帮助。
【关键词】先天畸形;并指(趾);遗传性疾病,先天性;遗传异质性
(J Int Reprod Health/Fam Plan,2015,35:170-176)
并指(趾)畸形(syndactyly,SD)是由于在肢体发育过程中,相邻的手指或脚趾没有分开造成的肢体畸形。作为最常见的一种遗传性肢体畸形,美国爱荷华州的发病率为0.03%~0.1%,在高加索人群中的发病率更是高达0.5%[1]。多呈常染色体显性遗传,另外有两个隐性遗传和一个X-连锁隐性遗传的表型。SD具有显著的临床异质性,可以累及单侧或双侧,可以呈对称或不对称畸形[2]。表型多变,在同一个体中会出现上下、左右肢体表型的不对称。SD可以分为部分并指或完全并指,皮肤融合性并指和骨性融合并指,可以仅涉及指骨,也可以进一步延伸至掌骨或跖骨,甚至到腕骨或跗骨水平。极轻度表型可能只是皮肤纹理的异常[3]。本文在Sajid Malik分类方案的基础上[4],汇总了近几年的遗传学研究成果。
1 并指(趾)畸形Ⅰ型
在所有已知的非综合征型的SD中,Ⅰ型并指最为常见。Ⅰ型并指表现为中轴织带:3/4指并指,和(或)2/3趾并趾。在发现几个并指(趾)家系中存在多个特征性表型和遗传变异后,又将Ⅰ型并指(趾)分为4个亚型。
1.1并指(趾)畸形Ⅰ-a型(Weidenreich型;zygodactyly;2/3脚趾并趾)这种常染色体显性遗传病最初被Weidenreich命名为zygodactyly。Zygodactyly是表现最不明显的一型,在临床实践中经常被忽视。其在男性的患病率约为4/10 000,占所有非综合征型SD病例的70%。其特征为2/3脚趾的皮肤性融合,双手正常(图1[4])。极少情况下,也会涉及其他脚趾,双脚表型通常一致。症状轻微的表型是2/ 3脚趾间的蹼轻微上升,或者只能通过皮纹异常检测才能发现。严重者,蹼可达到趾骨的顶端,甚至可以看到趾甲的紧密融合,第2脚趾倾斜内翻。Zygodactyly作为一个独特的遗传表型,其遗传学病因是由Malik等[5]在研究1个巴基斯坦大家系的遗传图谱时发现的。共分离实验显示Zygodactyly的致病基因位于染色体3p21~p31上的ZD1基因座(表1)。然而,德国家系的连锁分析数据提示,zygodactyly存在遗传异质性。
1.2并指(趾)畸形Ⅰ-b型(Lueken型;3/4并指和2/ 3并趾)这类亚型表现为3/4指和2/3趾的皮肤性融合(图1),有时也会存在以指端骨桥形式的骨融合,严重时可能累及第2到第5手指、第1到第5脚趾。在Bosse等[6]报告的家系中,显性表型定位到染色体2q34~q36上的SD1基因(表1)。

注:阴影部分表示皮肤融合;黑色阴影部分表示骨融合;虚线部分表示骨发育不良。图1Ⅰ~Ⅲ型并指(趾)畸形示意图[4]

表1并指(趾)畸形分类特征
1.3并指(趾)畸形Ⅰ-c型(Montagu型;3/4并指)这种罕见的常染色体显性遗传的特点是双手3/4手指的皮肤/骨性融合,双脚正常(图1)。Hsü[7]报道的1个中国家系中,23例受累者中显示了不同程度的双手3/4或3/4/5手指的骨性融合,只有1例患者存在3到5脚趾的部分融合。2014年Dai等[8]报道了两个中国人家系,通过连锁分析、基因测序,锁定致病基因为HOXD13(表1),并找到2个突变位点:p.R306Q和p. R306G。
1.4并指(趾)畸形Ⅰ-d型(Castilla型;4/5趾并趾)这种亚型表现为第4和第5脚趾的皮肤性融合,而且是已报道的第2常见的孤立性脚趾皮肤融合,发病率为0.22/万。各种轻微形式的皮肤性融合在临床实践中易被忽视。特别是由于错误的穿鞋方式,损坏了第5脚趾的外形。这一亚型的遗传模式和外显率尚少见报道。
2 并指(趾)畸形Ⅱ型[Vordingborg型;3/4手指和4/ 5脚趾并多指(趾)/趾;synpolydactyly;SPD]
Ⅱ型并指(趾)又称并多指(趾)(SPD),是临床上异质性最强的一类SD,呈常染色体显性遗传。SPD的标志性特征是3/4手指的皮肤性/骨性融合,第4和第5脚趾在并趾区域内的完全或部分的多趾(图1)。另外,SPD常伴有短指和屈曲指[9],是唯一有中轴多指/趾的SD。SPD具有显著的表型异质性,其所有的临床变异(约18种)可归结为3类:典型的SPD(A型);轻微变异的SPD(B型);罕见表型的SPD(C型),各分型的临床表型如图2所示[10-11]。SPD是外显率较低的显性遗传,目前已发现3个与SPD有关的基因座(SPD1~3)。SPD1临床和基因突变数据最完善,致病基因为HOXD13,典型的SPD1是由于HOXD13基因多聚丙氨酸延展突变所致,另外HOXD13基因的错义突变也是一大诱因[11-12]。有报道称FBLN1基因断裂可以导致SPD2[13]。通过连锁分析已经将SPD3的致病基因锁定在14q11.2~q13区域(表1),但是还未发现具体致病基因。
3 并指(趾)畸形Ⅲ型(Johnston-Kirby型;4/5或3/ 4/5并指)
该畸形影响4/5手指或3/4/5手指(图1)。小指的中节指骨发育不全,无名指为了适应与小指的融合,通常外翻,特别是完全融合的情况。并指通常内收,而且受影响的并指的指甲内侧通常也融合在一起。远端指节可能会形成骨桥,脚一般不受影响,遗传模式为不完全外显的常染色体显性遗传。

注:阴影部分表示皮肤融合;黑色阴影部分表示骨融合;虚线部分表示骨发育不良图2 SPD各临床表型(A、B、C)示意图[10]
4/5指并指,同时也是眼齿趾发育不良(ODDD)的一个特征,除此之外ODDD还有眼睛、耳朵等其他颜面特征。Schrander-Stumpel等[14]提出,ODDD和单纯的Ⅲ型并指都是由于邻近的基因缺失综合征引起的不同临床表型。分子生物学研究显示,ODDD和单纯的Ⅲ型并指都是由于编码连接蛋白的多效基因GJA1发生突变导致的(表1)[15-16]。
4 并指(趾)畸形Ⅳ型(Haas型;所有手指完全并指)
Haas型的并指患病率为0.033/万,呈常染色体显性遗传。表现为完全的皮肤性融合,伴有轴前或轴后多指。指甲可能为完全融合或只有轻微分离。手指的屈伸能力有限,而且手指连接在一起外观呈杯形。指骨有可能融为一个骨团,但是掌骨不存在骨性联结(图3)。
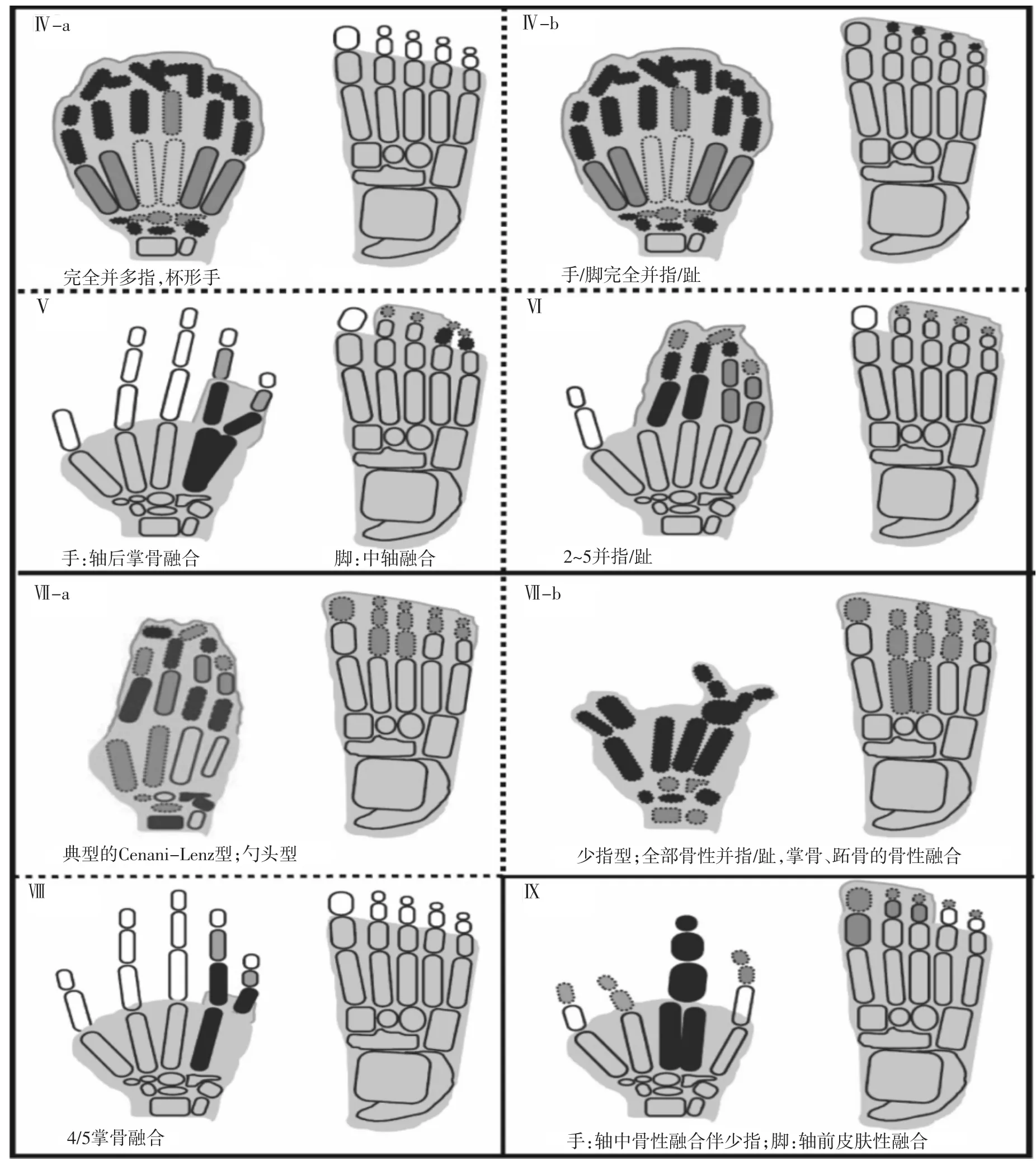
注:阴影部分表示皮肤融合;黑色阴影部分表示骨融合;虚线部分表示骨发育不良图3Ⅳ~Ⅸ型并指(趾)畸形示意图[4]
有文献报道,Ⅳ型并指(趾)存在两种变异体:①典型的Haas型并指,没有脚的参与;②除了手的完全融合外,还伴有5个脚趾的可变融合。Haas型并指已被证明是拇指多指畸形的另一种形式,只不过拇指多指畸形症状更轻。这两种表型都是由于染色体7q36(LMBR1基因)上的ZRS区域发生突变引起,该区域包含一个远程调节基因SHH[17]。Lohan等[18]的研究发现ZRS区域的微重复(≥80 kb)与Haas型并指有关(表1),而更小的微重复(<80 kb)则会导致更为严重的Laurin-Sandrow综合征。
5 并指(趾)畸形Ⅴ型(Dowd型;4/5掌骨的融合)
Ⅴ型的标志性特征是4/5掌骨的融合。其他症状包括融合的4/5掌骨的缩短,2到5手指的尺侧偏斜,3/4手指间的指叉,5手指的屈曲指,远端指骨的短指,以及受影响的手指远端指节间的折痕消失。脚部特征为:第1跖骨增生,第2至第5跖骨缩短,导致跖骨内翻,脚趾外翻(图3)。Ⅴ型SD属于常染色体显性遗传,通过对1个中国家系的研究,将其致病位点锁定为HOXD13基因的1个错义突变c.950A>G(表1)[19]。
6 并指(趾)畸形Ⅵ型(Mitten型;2/5指并指,2/5趾并趾)
Temtamy等[20]描述了1例并指患者右手从2至5手指融合在一起,而且远端指骨被合并为一个结状结构(图3),脚部表现为2/5并趾。该患者的第2个表弟也表现为同样的畸形,然而家系的其他成员则只有2/5脚趾的皮肤性融合,没有手部畸形。该SD为常染色体显性遗传,外显率低,且表型多变。
7 并指(趾)畸形Ⅶ型(Cenani-Lenz型,CLS;所有手指、脚趾严重的骨性融合,并伴有手部的变形)
这种常染色体隐性遗传的个体表现为严重的手、脚畸形。其特点是手骨排列紊乱,导致指骨无法识别。腕骨、掌骨和指骨的不规则骨性融合,给人以手被套在丝袜里的感觉(图3)。这种异常可能涉及桡骨和尺骨的融合、缩短或退化,导致桡骨脱位以及前臂缩短。下肢的变化类似于上肢,可能有些趾骨缺失。另外,某些罕见的患者可能累及颅面和肾脏功能。
Khan等[21]建议,Cenani-Lenz型并指分为两种截然不同的临床表型:(a)勺头型,(b)少指型。Kariminejad等[22]的研究显示勺头型和少指型包含或不包含肾脏畸形,都是由于参与Wnt/β信号通路的LRP4基因发生突变导致功能异常。另一项研究表明,非综合征型Cenani-Lenz样少指类表型,伴有肾功能缺损和听力损失,呈常染色体显性遗传是由于15q13.3区域的GREM1基因与FMN1基因发生重组引起的(表1)[23]。
8 并指(趾)畸形Ⅷ型(Orel-Holmes型;4/5掌骨的融合;X-连锁隐性遗传)
该类型的特征为4/5掌骨融合,小指尺侧偏斜,无其他异常(图3)。4/5掌骨缩短,使得远端的指骨过度分离,无法与其他手指平行,遗传模式为X-连锁隐性遗传或常染色体显性遗传。2013年,Jamsheer等[24]通过对1个波兰家系和1个德国家系进行外显子测序时发现,位于Xq21.1区域的FGF16基因的两个无义突变p.R179X和p.S157X导致Ⅷ型SD。2014年该学者在1个散发男性病例中发现FGF16基因的1个截短突变p.E158DfsX25,进一步证实FGF16基因与肢体发育有关(表1)[25]。
9 并指(趾)畸形Ⅸ型(Malik-Percin型;3/4掌骨融合,伴有中轴少指,轴前并趾)
Percin等[26]和Malik等[27]等分别描述了1个土耳其和巴基斯坦的近亲家系,其中受影响的个体表现为中轴少指,3/4掌骨融合为一个掌骨,拇指畸形,小指发育不全和屈曲指(图3),另外伴有轴前并趾,而且所有的脚趾趾骨发育不全。由于其并没有延伸至腕骨/跗骨,而且也不是以骨骼的异常排列为特征,临床上其严重程度要弱于CLS。该SD为常染色体隐性遗传,致病基因定位于染色体17p13.3(表1)。
10 结语
SD是肢体发育的过程中指/趾节没有有效分离造成的。作为1个明确的表型,孩子一出生便引起医生的注意,尤其是出现在上肢时。脚比手更易受影响,而且男性的发病率是女性的2倍[28]。
SD的分型是相当令人困扰的,例如,家庭内个体间表型存在变异性,既有相同的表型,也有差异表型,这就给分型工作带来麻烦。尤其是只有极少数个体存在特定的表型,而其他个体是罕见的SD(如:缺指畸形,SPD,短指畸形,屈曲指畸形)时,分型工作更是令人望而生畏。很多因素如:遗传模式,不完全外显率,遗传异质性,大量的成形素参与肢体发育,成形素之间的相互作用,以及修饰基因或远程调控基因的参与(例如ZRS)都会使分型工作复杂化。因此,即使充分考虑肢体形态和分子胚胎学进展,仍无法将特定的SD与对应基因进行关联。目前这个分类方案是结合临床、遗传和分子生物学进展而制定的。
SD是研究造成并指(趾)家系个体间临床异质性的各种因素如:遗传机制、表观遗传学、多效性、随机性因素之间关系的良好模型。随着SD遗传学数据的逐渐完善、产前诊断技术的发展,利用羊水进行基因测序可以诊断大部分的严重SD的胎儿,降低新生儿出生缺陷的发生。
参考文献
[1] Jordan D,Hindocha S,Dhital M,et al. The epidemiology, genetics and future management of syndactyly [J]. Open Orthop J,2012,6:14-27.
[2] Deng H,Tan T. Advances in the Molecular Genetics of Non -syndromic Syndactyly [J]. Curr Genomics,2015,16(3):183-193.
[3] Chopra K,Tadisina KK,Patel KR,et al. Syndactyly repair [J]. Eplasty,2013,13:ic51.
[4] Malik S. Syndactyly: phenotypes, genetics and currentclassification [J]. Eur J Hum Genet,2012,20(8):817-824.
[5] Malik S,Schott J,Ali SW,et al. Evidence for clinical and genetic heterogeneity of syndactyly type I: the phenotype of second and third toe syndactyly maps to chromosome 3p21.31 [J]. Eur J Hum Genet,2005,13(12):1268-1274.
[6] Bosse K,Betz RC,Lee YA,et al. Localization of a gene for syndactyly type 1 to chromosome 2q34-q36 [J]. Am J Hum Genet,2000,67(2):492-497.
[7] Hsü CK. Hereditary syndactylia in a Chinese family [J]. Chin Med J,1965,84(7):482-485.
[8] Dai L,Liu D,Song M,et al. Mutations in the homeodomain of HOXD13 cause syndactyly type 1-c in two Chinese families[J]. PLoS One,2014,9(5):e96192.
[9] Brison N,Debeer P,Tylzanowski P. Joining the fingers: a HOXD13 Story[J]. Dev Dyn,2014,243(1):37-48.
[10] Malik S,Grzeschik KH. Synpolydactyly: clinical and molecular advances [J]. Clin Genet,2008,73(2):113-120.
[11] Wang B,Xu B,Cheng Z,et al. A novel non-synonymous mutation in the homeodomain of HOXD13 causes synpolydactyly in a Chinese family [J]. Clin Chim Acta,2012,413(13/14):1049-1052.
[12] Shi X,Ji C,Cao L,et al. A splice donor site mutation in HOXD13 underlies synpolydactyly with cortical bone thinning [J]. Gene,2013,532(2):297-301.
[13] Debeer P,Schoenmakers EF,Twal WO,et al. The fibulin-1 gene (FBLN1) is disrupted in a t (12;22) associated with a complex type of synpolydactyly [J]. J Med Genet,2002,39(2):98-104.
[14] Schrander-Stumpel CT,De Groot-Wijnands JB,De Die-Smulders C,et al. Type III syndactyly and oculodentodigital dysplasia: a clinical spectrum[J]. Genet Couns,1993,4(4):271-276.
[15] Jamsheer A,Sowińska -Seidler A,Socha M,et al. Three novel GJA1 missense substitutions resulting in oculo -dento -digital dysplasia (ODDD) - further extension of the mutational spectrum [J]. Gene,2014,539(1):157-161.
[16] Nishat S,Mansoor Q,Javaid A,et al. Oculodentodigital Syndrome with Syndactyly Type III in a Pakistani consanguineous family [J]. J Dermatol Case Rep,2012,6(2):43-48.
[17] Dai L,Guo H,Meng H,et al. Confirmation of genetic homogeneity of syndactyly type IV and triphalangeal thumb -polysyndactyly syndrome in a Chinese family and review of the literature [J]. Eur J Pediatr,2013,172(11):1467-1473.
[18] Lohan S,Spielmann M,Doelken SC,et al. Microduplications encompassing the Sonic hedgehog limb enhancer ZRS are associated with Haas -type polysyndactyly and Laurin -Sandrow syndrome [J]. Clin Genet,2014,86(4):318-325.
[19] Zhao X,Sun M,Zhao J,et al. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome [J]. Am J Hum Genet,2007,80(2):361-371.
[20] Temtamy SA,McKusick VA. The genetics of hand malformations [J]. Birth Defects Orig Artic Ser,1978,14(3):i-xviii,1-619.
[21] Khan TN,Klar J,Ali Z,et al. Cenani-Lenz syndrome restricted to limb and kidney anomalies associated with a novel LRP4 missense mutation [J]. Eur J Med Genet,2013,56(7):371-374.
[22] Kariminejad A,Stollfuβ B,Li Y,et al. Severe Cenani -Lenz syndrome caused by loss of LRP4 function[J]. Am J Med Genet A,2013,161A(6):1475-1479.
[23] Dimitrov BI,Voet T,De Smet L,et al. Genomic rearrangements of the GREM1 -FMN1 locus cause oligosyndactyly, radio -ulnar synostosis, hearing loss, renal defects syndrome and Cenani --Lenz-like non-syndromic oligosyndactyly[J]. J Med Genet,2010,47(8):569-574.
[24] Jamsheer A,Zemojtel T,Kolanczyk M,et al. Whole exome sequencing identifies FGF16 nonsense mutations as the cause of X-linked recessive metacarpal 4/5 fusion[J]. J Med Genet,2013,50(9):579-584.
[25] Jamsheer A,Smigiel R,Jakubiak A,et al. Further evidence for FGF16 truncating mutations as the cause of X-linked recessive fusion of metacarpals 4/5[J]. Birth Defects Res A Clin Mol Teratol,2014,100(4):314-318.
[26] Percin EF,Percin S,Egilmez H,et al. Mesoaxial complete syndactyly and synostosis with hypoplastic thumbs: an unusual combination or homozygous expression of syndactyly type I?[J]. J Med Genet,1998,35(10):868-874.
[27] Malik S,Arshad M,Amin -Ud -Din M,et al. A novel type of autosomal recessive syndactyly: clinical and molecular studies in a family of Pakistani origin[J]. Am J Med Genet A,2004,126A(1):61-67.
[28] Mandarano -Filho LG,Bezuti MT,Akita R,et al. Congenital syndactyly: case by case analysis of 47 patients[J]. Acta Ortop Bras,2013,2(6):333-335.
[本文编辑王琳]
LIU Jin-xiu,CHEN Wei,WANG Xiang-rong,ZHOU Qing,SI Biao,DUAN Wen-yuan. Institute of Cardiovascular Disease,General Hospital of Jinan Military Region,Jinan 250022,China
【Abstract】Syndactyly (SD) is one of the most common hereditary limb malformations depicting the fusion of certain fingers or toes. It may occur as an isolated entity or a component of more than 300 syndromic anomalies. Syndactylies exhibit great inter- and intra-familial clinical variability. Phenotype can be unilateral or bilateral and symmetrical or asymmetrical within a subject. At least nine non-syndromic syndactylies have been characterized. Most of the syndactyly types are inherited as autosomal dominant but two autosomal recessive and an X-linked recessive entity have also been described. Whereas the underlying genes/mutations for typesⅡ-a, Ⅲ,Ⅳ,Ⅴ,ⅦandⅧhave been worked out, the etiology and molecular basis of the other syndactyly types remain unknown. In this communication, based on the classification of Sajid Malik, we summarized the results of genetic studies on syndactyly in recent years, which will contribute to further investigation of the pathogenic mechanism and implementation of genetic diagnosis of syndactyly.
【Keywords】Congenital abnormalities;Syndactyly;Genetic diseases,inborn;Genetic heterogeneity
收稿日期:(2015-11-11)
Corresponding author:DUAN Wen-yuan,E-mail:dwy2115@126.com
通信作者:段文元,E-mail:dwy2115@126.com
基金项目:国家重点基础研究规划(2013CB945402)
作者单位:250022济南军区总医院心血管病研究所
Syndactyly: Classification and Genetics
