PLK1 PBD靶向抗肿瘤抑制剂的筛选及活性研究
2016-03-29宗可昕张晶陈云雨范卓文司书毅
宗可昕,张晶,陈云雨,范卓文,司书毅
PLK1 PBD靶向抗肿瘤抑制剂的筛选及活性研究
宗可昕*,张晶*,陈云雨,范卓文,司书毅
【摘要】
目的筛选靶向 PLK1 PBD 的抗肿瘤小分子抑制剂,对其抗肿瘤效果进行体外评价,为抗肿瘤药物提供先导化合物。方法利用荧光偏振模型初筛 PLK1 PBD 抑制剂;通过MTT 法寻找初筛阳性化合物的敏感细胞系;利用流式细胞仪检测化合物对细胞周期和凋亡的影响;通过分子对接技术探讨化合物与 PLK1 PBD 结合方式;利用划痕实验测定化合物对肿瘤细胞迁移的影响。
结果从 20 000 个化合物中筛选得到活性化合物 1097C11,MTT 结果显示其能抑制多种肿瘤细胞系的增殖;流式细胞仪检测其能够促进肿瘤细胞凋亡,在 25 μmol/L 时,晚期凋亡达到 77.02%;能导致细胞G1/M 期阻滞,分子对接结果显示 1097C11 与 PLK1 PBD 结构域具有良好的亲和性,细胞迁移结果显示 1097C11 能够抑制 HCT-116 细胞的迁移,在 20 μmol/L时,迁移率低至 33.4%。
结论化合物 1097C11 具有较好的抗肿瘤活性,并有望成为靶向 PLK1 PBD 的抗肿瘤先导化合物。
【关键词】荧光偏振;抗肿瘤药;保罗样激酶 1 抑制剂
www.cmbp.net.cn中国医药生物技术, 2016, 11(1):13-20
作者单位:150040 哈尔滨,黑龙江中医药大学药学院药分教研室(宗可昕、范卓文);100050 北京,中国医学科学院医药生物技术研究所国家新药(微生物)筛选实验室(宗可昕、张晶、陈云雨、司书毅);130022长春,中国科学院长春应用化学研究所(陈云雨)
*同为第一作者
近 20 年,恶性肿瘤在我国人口死因中所占的比例与日俱增,现今虽然有很多抗肿瘤药物,尚存在选择性差、毒副作用强、易产生耐药性等缺点,靶向抗肿瘤小分子抑制剂可以达到高选择性、低毒性的治疗效果,从而克服传统药物的缺点,使抗肿瘤药物研究进入了一个崭新的阶段。
细胞的周期是由不同激酶调控的,其中保罗样激酶(polo-like kinases,PLKs)是广泛存在于真核生物中的由丝/苏氨酸激酶结构域的 N 端和具有调节 PLKs 激酶活性及亚细胞动态定位的特征性的 C 端结构域组成的一类高度保守的丝/苏氨酸蛋白激酶[1-3],越来越引起人们的重视。PLK 家族成员包括 PLK1、PLK2、PLK3、PLK4 和 PLK5。家族不同成员的 C 端特征性 PBD 结构域对同一底物的亲和力有很大的区别[4]。PLK1 在大部分恶性肿瘤细胞中呈过度表达并且与肿瘤的发生与发展密切相关,临床数据表明通过抑制 PLK1 能阻止体外肿瘤细胞的扩散,抑制细胞增殖与促进细胞凋亡[5-7]。PLK1 主要在细胞有丝分裂的 G2 晚期和M 期表达,在有丝分裂启动、中心体成熟、纺锤体组装、染色体分离及胞质分裂等细胞有丝分裂过程中发挥至关重要的作用[8-14]。
荧光偏振法是一种被广泛利用在临床和生物医学领域的快速和定量分析不同分子和酶相互作用的技术[15-17],20 世纪 90 年代中期开始应用于药物的高通量筛选,并且已经成为研究蛋白质-蛋白质、蛋白质-DNA、DNA-DAN、配体-受体、抗原-抗体等相互作用的有力工具。近年来,在以蛋白质-蛋白质相互作用(protein-protein interactions,PPIs)为靶点的药物高效发现研究中展现出无可比拟的应用前景。
本研究通过对中国医学科学院医药生物技术研究所国家新药微生物筛选中心样品库中筛选出来的编号为 1097C11 的化合物的抗肿瘤分子机制进行初步发掘,探讨其辅助治疗恶性肿瘤的可能性。
1 材料与方法
1.1材料
1.1.1化合物和细胞20 000 个待筛化合物购自北京百灵威科技有限公司;人结直肠腺癌细胞HT-29 用 DMEM/F12 + 5% FBS + NEAA(1:100)培养基培养;乳腺癌细胞 MCF-7、人宫颈癌细胞
HeLa、人结直肠腺癌细胞 HCT-116 和人正常肝细胞系 L02 均用含 10% FBS 的 DMEM 培养基培养;人成骨肉瘤细胞 MG-63 和人肝癌细胞 HepG2用含 10% FBS 的 MEM 培养基培养;人前列腺癌细胞 PC3 用含 10% FBS 的 F12k 培养基培养;人肺癌细胞 A549 用含 10% FBS 的 F12 培养基培养,以上细胞均由中国医学科学院医药生物技术研究所重点实验室提供。
1.1.2试剂FITC 标记多肽由上海强耀生物科技有限公司合成;重组蛋白PLK1 PBD 由陈云雨博士提供;NaCl、Tris-HCl、EDTA、RNaseA、碘化丙啶(PI)、噻唑蓝(MTT)、DMSO 均购自北京普利莱基因技术有限公司;Annexin V-FITC/PI 双染试剂盒购自北京四正柏生物科技有限公司。
1.1.3仪器Envision 2014 multilabel reader 酶标仪为美国 PerkinElmer 公司产品;FACS caliur 型流式细胞仪为美国 Beckman Coulter 公司产品;WD-9405B 型水平摇床为北京市六一仪器厂产品;DFC450C 型活细胞显微观察及图像采集仪为德国Leica 公司产品。
1.2方法
1.2.1化合物初筛利用荧光偏振模型对 1 mg/ml的化合物库进行初步筛选。将400 nmol/L PLK1 PBD 溶液以 30 μl/孔依次加入到 384 孔板中,再将供筛选的化合物以 0.3 μl/孔依次加入,以加入0.3 μl DMSO 孔作为阴性对照,不加 PLK1 PBD的为阳性对照,将其混匀后室温避光 140 r/min 振摇,孵育 60 min 后加入 30 μl/孔 60 nmol/L 的FITC-Probe,继续避光孵育 15 min,以多功能微孔板检测仪进行样品的偏振值检测并进行数据分析。化合物抑制率按如下公式计算:
抑制率(%)=(1 – OD受试药组– OD空白组)× 100% OD正常组– OD空白组
1.2.2小分子抑制剂的量效关系分析将初筛阳性化合物 1097C11 溶于 DMSO 中,配置成10 mmol/L 的母液。母液倍比稀释,按照初筛的方法在 FITC 的模型上检测,以浓度的对数为横坐标,抑制率为纵坐标,利用 Graphpad prism5 软件分析计算 IC50值[18-19]。
1.2.3MTT 比色法测定化合物在细胞上的杀伤作用复苏细胞,培养至对数生长期,接种于 96 孔板中,控制每孔的细胞量为 2 × 104个/ml。24 h 后,弃掉培养基,将化合物倍比稀释后,以 100 μl/孔加入 96 孔板中,3 个复孔,不加化合物组为空白对照组,继续培养 48 h。每孔加入终浓度为 0.5 mg/ml 的 MTT 避光孵育 4 h 后弃掉培养基,加入 150 μl DMSO 避光振荡 15 min,酶标仪 570 nm检测[20]。数据处理方法同 1.2.2。计算 IC50值。抑制率计算方式如下:
抑制率(%)=(1 –1 – OD受试药组)× 100% OD空白组平均值
1.2.4流式细胞仪检测细胞凋亡利用 Annexin V-FITC/PI 双染法测定细胞凋亡,将培养至对数期的 HCT-116 细胞以每孔 1 × 105个/ml 接种于六孔板中[21],待 24 h 进入对数生长期后加入浓度为0、5、15、25 μmol/L 的 1097C11 作用 24 h,取出上述细胞培养板,以 PBS 小心漂洗 1 次,再以0.25% 胰酶消化细胞,1000 r/min 离心 5 min,收集细胞。将上述消化的 HCT-116 细胞以 4 ℃ 预冷的 PBS 洗涤 2 次,用 250 μl 1 × 结合缓冲液悬浮细胞,调节其浓度为 5 × 106个/ml。再加入 5 μl Annexin V-Alexa Fluor488,室温避光孵育 30 min,再加入 5 μl PI,室温避光孵育 5 ~ 10 min。将上述细胞悬液过滤到 BD 流式管中,补加 400 μl PBS并做好对应的标记,以流式细胞仪进行检测和数据统计分析。
1.2.5细胞同步化及 FACS 分析化合物对周期的影响将生长到对数生长期的 HCT-116 细胞以2 × 105个/ml 接种于六孔板中,待细胞贴壁后加入浓度为 0.1 mol/L 的胸腺嘧啶 40 μl 培养 8 ~ 12 h后,用 PBS 洗 3 次,换成正常培养基继续培养12 h 后,再次加入 0.1 mol/L 的胸腺嘧啶 40 μl 培养 8 ~ 12 h,PBS 洗过 3 次后加入浓度为 0、5、15、25 μmol/L 的化合物 1097C11。作用 18 h 后,取出上述细胞培养板,以 PBS 小心漂洗 1 次,再以 0.25% 胰酶消化细胞,1000 r/min 离心 5 min,收集细胞,用 PBS 洗净,用 70% 冰乙醇 4 ℃ 固定 1 h 以上(通常过夜),PBS 洗净乙醇,加入终浓度为 100 μg/ml 的 RNaseA 和终浓度为 50 μg/ml 的 PI,再补加 PBS 至 500 μl 体系,于 37 ℃ 水浴锅中避光孵育 30 min。1 h 内过滤,上流式细胞仪进行数据统计分析。
1.2.6化合物 1097C11 和 PLK1 蛋白的 PBD结构域的对接采用 Discovery Studio4.0 软件进行分子对接。从蛋白质数据库(protein data bank,PDB)中下载 PLK1 PBD 的 PDB(4H71)文件,进行预处理,去水,加氢,处理蛋白,确定活性位点,将 1097C11 与活性位点对接。
1.2.7细胞划痕实验先用记号笔在六孔板背面均匀划得横线,大约每隔 0.5 ~ 1 cm 横穿过孔,每孔至少穿过三条线,以便获得不同视野。以 5 × 105个/ml 接种 HCT-116 细胞于六孔板中。第 2 天用无菌枪头比着直尺,尽量垂直于背面的横线划痕。用 PBS 洗细胞 3 次,除去划线下的细胞,加入含 5、15、20 μmol/L 的 1097C11。放入 37 ℃、5% CO2的培养箱孵育培养 48 h 后取样,拍照。按照以上方法用 20 μmol/L 的 1097C11 作用于HCT-116 细胞不同时间取样拍照。
2 结果
2.1化合物初次筛选结果
利用荧光偏振的方法对化合物库中的 20 000 个化合物进行筛选,最终得到活性化合物1097C11,对 PLK1 PBD 的抑制率较高,达到 90% 以上,经过 ChemDraw ISIS查询到化合物结构(图1)。

图1 化合物 1097C11 的结构式Figure 1 The structure of compound 1097C11
2.2阳性化合物 1097C11 在 PLK1 PBD 上的活性
从国际经验看,市场化定价是成品油市场最普遍采用的模式。韩国的成品油价格市场化进程经历了由政府定价过渡为与国际市场接轨再到价格完全市场化三个阶段,最终形成了既能够反映国际市场变化,又能体现国内市场实际供需情况的价格体系。明确合理的价格运行机制,不仅能够提高成品油市场运行效率,也有利于保障国家能源供应安全,提升国内石油企业竞争力。
将化合物 1097C11 由 10 μg/ml 二倍稀释 8 个浓度,利用软件 Graphpad prism5 拟合曲线,可看到随着 1097C11 浓度的增高,对 PLK1 PBD 活性的抑制率逐渐增强,最终趋于平稳,具有明显的剂量依赖性。如图2 所示,1097C11 对 PLK1 PBD 的 IC50约为 1.78 μmol/L。
2.3化合物 1097C11 对细胞增殖的抑制作用
将化合物 1097C11 从 100 μmol/L 二倍稀释至1.56 μmol/L,作用于以下几种细胞系,经过 Graphpad prism5 软件分析计算,IC50见表1。从表1 中我们可以发现,1097C11 对 HCT-116 的增殖抑制作用最强,IC50值为 8.43 μmol/L(图3)。而在正常肝细胞 L02 上的 IC50值为 10.15 μmol/L,在体外对肿瘤细胞和正常细胞毒性相差不大,选择性较差。而根据文献报道,PLK1 PBD 在 HCT-116 中的蛋白表达量较高[22],所以选择 HCT-116 用来做后续试验。

图2 1097C11 对体外纯化 PLK1 PBD 的抑制率Figure 2 Inhibition ratio of 1097C11 against purified PBD in vitro

表1 1097C11 对不同细胞株的毒性Table 1 Cytotoxicity of 1097C11 to different cells
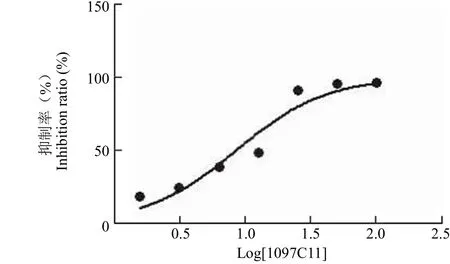
图3 1097C11 在 HCT-116 细胞中的抑制率Figure 3 Inhibition ratio of 1097C11 for HCT-116 cell
2.4化合物 1097C11 对细胞凋亡的影响
在 AnnexinⅤ/PI 双染实验中,0、5、15、25 μmol/L 的小分子抑制剂1097C11 作用 HCT-116细胞 24 h 后,通过 FACS 检测,与 DMSO 对照组相比,随着 1097C11 剂量的不断增加,HCT-116细胞的凋亡率逐渐升高,晚期凋亡细胞数目随着剂量的不断增加而增多并有早期凋亡,在浓度达到 25 μmol/L 时晚期凋亡细胞数达到总细胞数的77.02%,如图4、图5 所示。
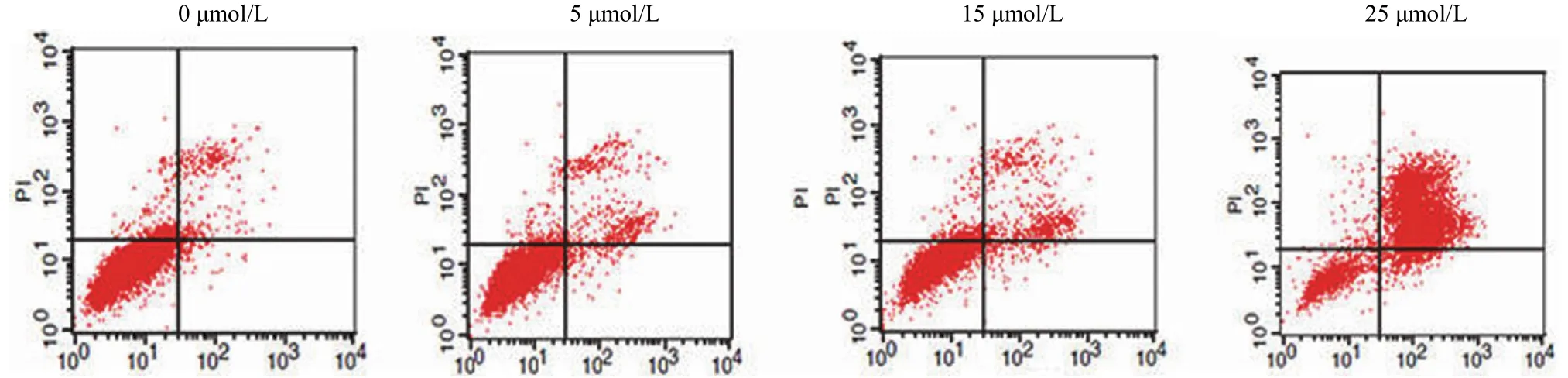
图4 1097C11 对 HCT-116 细胞的促凋亡作用Figure 4 Effect of compound 1097C11 on apoptosis in HCT-116 cells
2.5化合物 1097C11 对细胞周期的阻滞作用
结果表明,1097C11 可以明显地使周期同步化的 HCT-116 细胞发生 G2/M 期阻滞,延缓细胞周期的进程。利用细胞周期同步化的技术获得周期一致性的细胞,提高了结果的准确性和可靠性。当 5、15、25 μmol/L 的 1097C11 作用于周期同步化的HCT-116 细胞 24 h 时,与对照组相比,随着剂量的不断增加,HCT-116 细胞的 G2/M 期比率逐渐升高,有很强的剂量依赖性。对照组 G2/M 期比率为 9.84%,当剂量达到 25 μmol/L 时,G2/M 期比率达到 44.03%(图6)。
PBD 由 PB1 和 PB2 通过构象的转变在空间上形成二聚体,依靠氢键、范德华力及疏水键形成一个“拉链”式结构,磷酸肽底物的结合区域恰好位于 PB1 和 PB2 之间的“狭窄沟槽”内。分子对接中,我们发现小分子抑制剂 1097C11 与水化His538、ASN533 和 Lys540 的侧链形成较为牢固的氢键;同时与 Arg557 形成叠加的 π 键,进而使 1097C11/PBD 复合物更加稳定(图7)。表明小分子抑制剂 1097C11 与 PLK1 PBD 的非共价键的竞争结合方式,但是这种过于“紧密”的结合模式的精确解析还有待其结构生物学的深入研究。
2.71097C11 对 HCT-116 细胞迁移的影响
HCT-116 细胞迁移实验结果显示,与对照组相比,加药组 HCT-116 细胞的迁移能力显著减弱,尤其是在 20 μmol/L 时,加药组的细胞迁移能力和对照组比有明显差异,表明该化合物能够抑制HCT-116 细胞的迁移,并且可以看出随着 1097C11浓度的增加,这种剂量依赖关系也显著增加。而20 μmol/L 的 1097C11 在不同作用时间表现出不同的迁移状态,与对照组相比随着时间的延长,加药组的迁移抑制作用 48 h 比 12 h 时迁移减弱的最明显,表现出一定的时间依赖趋势(图8 和图9)。

图5 1097C11 对 HCT-116 细胞的促凋亡作用Figure 5 Effect of compound 1097C11 on apoptosis in HCT-116 cells
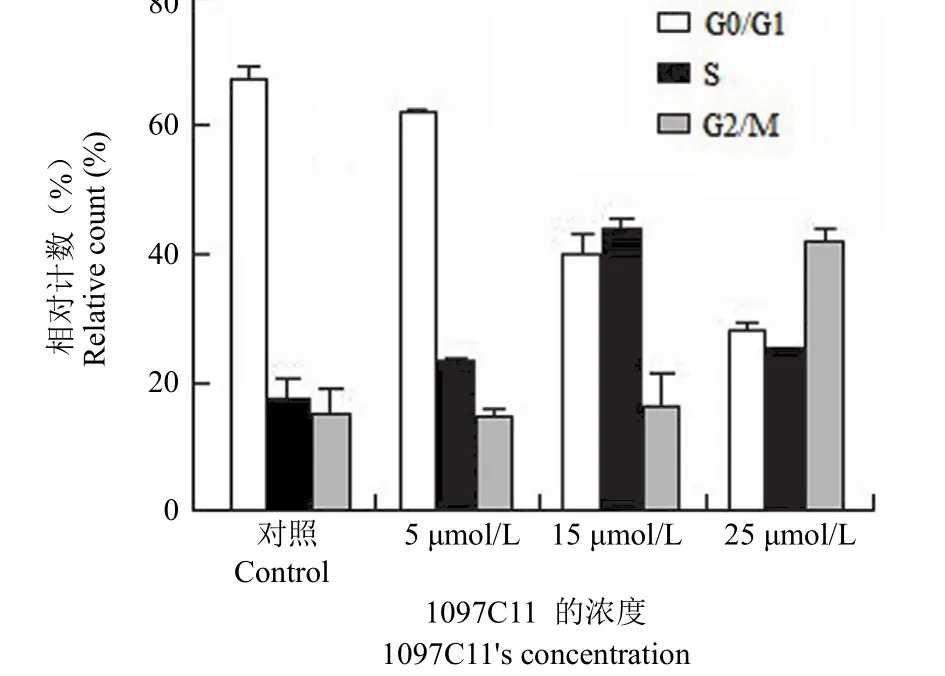
图6 1097C11 对 HCT-116 细胞的周期阻滞作用Figure 6 Effect of compound 1097C11 on cell cycle in HCT-116 cells

图7 1097C11 与 PBD1 活性区域的虚拟对接(A:小分子蛋白相互作用二维平面图;B:蛋白小分子相互作用图;C:蛋白与小分子空间位置示意图)Figure 7 Virtual docking of 1097C11 with PBD1 domain (A: 2D floor plan of the interaction of the protein and small molecule; B: Interaction diagrams of the protein and small molecule; C: Spatial location schematic diagram of the protein and small molecule)

图8 1097C11 对 HCT-116 的迁移影响Figure 8 Effect of 1097C11 on migration ability in HCT-116 cells

图9 1097C11 不同作用时间对 HCT-116 的迁移影响Figure 9 Effects of 1097C11 on migration ability at different time intervals in HCT-116 cells
3 讨论
临床前期试验显示,PLK1 是多种肿瘤治疗的有效靶点,PLK1 蛋白表达水平与肿瘤细胞的增殖有关,通过不同途径调节细胞周期的启动[23],而以PLK1 为靶点的药物较传统微管蛋白活性抑制剂(如紫杉醇、长春碱、长春瑞宾)具有良好的临床优势,故对于 PLK1 PBD 靶点的小分子抑制剂的研究是很有临床意义的。
荧光偏振方法 1926 年由 Perrin 提出,其原理在于:荧光团的极化程度是与分子旋转速度逆相关的,当小的荧光分子在平面极化光的激发下,在荧光寿命内(激发与发射之间的时差)小分子在溶液中的旋转速度快,所形成的发射光大部分是去极化的,偏振值较低;但是,当荧光分子结合到大分子后,它的有效体积增大,旋转减慢,偏振值较高。从而提供了直接检测荧光分子结合到受体比值的方法[24-26]。该法不仅价格低廉而且速度快、效率高、操作简单。本研究利用荧光偏振模型对中国医学科学院医药生物技术研究所国家新药微生物筛选中心样品库的 20 000 个化合物进行筛选,最终选出活性较好的化合物 1097C11,经查询,该化合物是个新型结构的抗肿瘤化合物。通过荧光偏振模型得到小分子抑制剂 1097C11 的 MTT 结果发现,1097C11 对结肠癌细胞 HCT-116 作用较为明显,这可能与靶蛋白在不同细胞中的表达量有关。其在正常肝细胞中选择性不高,但有报道证实,肿瘤组织 PLK1 蛋白表达明显高于正常肝组织中的PLK1 蛋白表达量,人类多数疾病并不是由单一基因或靶点导致的疾病,化合物靶标并不是疾病所独有的,多数具有多重生物功能,或者与其他功能蛋白相互影响也会导致选择性低,同时多靶点抗癌药物不仅可以提高治疗效果,而且有可能避免多种药物联合用药而导致的药物相互作用,降低副作用,在癌症治疗中具有重要的意义[27-29]。
流式细胞仪检测结果表明,1097C11 能够很明显地促进细胞凋亡,在 25 μmol/L 浓度下,晚期凋亡达到 77.02% 左右,而在细胞周期中,1097C11对于 G1/M 期的阻滞作用也有一定的量效关系,这也与之前的报道相符合[30],表明小分子抑制剂1097C11 对 PLK1 PBD 有着较为明显的抑制作用。分子对接是分子模拟的重要方法之一,其本质是两个或多个分子之间的识别过程,其过程涉及分子之间的空间匹配和能量匹配[31-33],通过蛋白和小分子结合的位点判断小分子抑制剂和蛋白的结合能力。经过分子对接进一步证实了小分子抑制剂1097C11 与 PBD 结合,进而起到抑制作用,因而证明 1097C11 是以 PBD 为作用靶点发挥抗肿瘤作用。同时细胞迁移实验检测结果也表明,1097C11可以抑制 HCT-116 细胞的迁移,并呈现一定的时间和浓度依赖关系。
综上所述,本研究表明小分子抑制剂 1097C11有较好的 PBD 靶向抗肿瘤活性,这为以后开发安全、高效的 PLK1 PBD 小分子抑制剂奠定了坚实的理论基础。随着基础研究及临床研究的不断深入,以 PLK1 PBD 为靶点的小分子抑制剂必将成为肿瘤靶向治疗和个体化治疗策略的新突破口。
参考文献
[1] Viala M, Brosseau S, Planchard D, et al. Second generation ALK inhibitors in non-small cell lung cancer: systemic review. Bull Cancer, 2015, 102(4):381-389.
[2] Lozano-Blázquez A, Dickson R, Fraga-Fuentes MD, et al. Differences in cancer drug assessment between Spain and the United Kingdom. Eur J Cancer, 2015, 51(13):1843-1852.
[3] Fenton B, Glover DM. A conserved mitotic kinase active at late anaphase-telophase in syncytial Drosophila embryos. Nature, 1993, 363(6430):637-640.
[4] Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol, 2009, 10(4):265-275.
[5] Kothe M, Kohls D, Low S, et al. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry, 2007, 46(20):5960-5971.
[6] Zhou Y, Tang YL, Gan RL, et al. The research development of PLK1 in tumorigenesis. J Med Res, 44(3):158-161. (in Chinese)
周燕, 唐运莲, 甘润良, 等. PLK1在肿瘤发生中的研究进展. 医学研究杂志, 2015, 44(3):158-161.
[7] Hafner M, Vianini E, Albertoni B, et al. Displacement of protein-bound aptamers with small molecules screened by fluorescence polarization. Nat Protoc, 2008, 3(4):579-582.
[8] Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, et al. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol, 2005, 15(24):2199-2207.
[9] Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol, 2009, 4(10):265-275.
[10] Garuti L, Roberti M, Bottegoni G. Polo-like kinases inhibitors. Curr Med Chem, 2012, 19(23):3937-3948.
[11] de Cárcer G, Manning G, Malumbres M. From Plk1 to Plk5: functional evolution of polo-like kinases. Cell Cycle, 2011, 10(14):2255-2262.
[12] Caruso M, Valsasina B, Ballinari D, et al. 5-(2-amino-pyrimidin-4-yl)-1H-pyrrole and 2-(2-amino-pyrimidin-4-yl)-1,5,6,7-tetrahydropyrrolo[3,2-c]pyridin-4-one derivatives as new classes of selective and orally available Polo-like kinase 1 inhibitors. Bioorg Med Chem Lett, 2012, 22(1):96-101.
[13] Shan HM, Shi Y, Quan J. Identification of green tea catechins as potent inhibitors of the polo-box domain of polo-like kinase 1. Chem Med Chem, 2015, 10(1):158-163.
[14] Qian WJ, Park JE, Lim D, et al. Peptide-based inhibitors of Plk1 polo-box domain containing mono-anionic phosphothreonine esters and their pivaloyloxymethyl prodrugs. Chem Biol, 2013, 20(10):1255-1264.
[15] Jameson DM, Croney JC. Fluorescence polarization: past, present and future. Comb Chem High Throughput Screen, 2003, 6(3):167-173.
[16] Nasir MS, Jolley ME. Fluorescence polarization: an analytical tool for immunoassay and drug discovery. Comb Chem High Throughput Screen, 1999, 2(4):177-190.
[17] Lea WA, Simeonov A. Fluorescence polarization assays in small molecule screening. Expert Opin Drug Discov, 2011, 6(1):17-32.
[18] Cer RZ, Mudunuri U, Stephens R, et al. IC50-to-Ki: a web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res, 2009, 37(Web Server issue):W441-W445.
[19] Feng WZ, Lin JH, Cai SL, et al. Rapid and high throughput measurement of lipase thermo-stability through ANS fluorescence signal assay. Chin J Biotech, 2011, 27(4):584-591. (in Chinese)
冯蔚宗, 林俊涵, 菜少丽, 等. 高通量脂肪酶稳定性测定法-ANS法的建立. 生物工程学报, 2011, 27(4):584-591.
[20] Liu M, Zhang Y, Liao Y, et al. Evaluation of the antitumor efficacy of RNAi-mediated inhibition of CDC20 and heparanase in an orthotopic liver tumor model. Cancer Biother Radiopharm, 2015, 30(6):233-239.
[21] Zhu H, Deng K, Zhao YQ, et al. The effects of asmase mediated endothelial cell apoptosis in multiple hypofractionated irradiations in CT26 tumor bearing mice. Asian Pac J Cancer Prev, 2015, 16(11): 4543-4548.
[22] Gao JY, Yu SH, Zhao YH, et al. Expression and significance of PLK1 and STK15 gene, and effect of its specific inhibitor on proliferation in colon cancer cells. Chin J Bases Clin General Surg, 2014, 21(6):702-706. (in Chinese)
高俊勇, 余少鸿, 赵永恒, 等. PLK1及STK15基因在结肠癌细胞中的表达及其特异性抑制剂对结肠癌细胞增殖的影响. 中国普外基础与临床杂志, 2014, 21(6):702-706.
[23] Reindl W, Yuan J, Krämer A, et al. Inhibition of polo-like kinase 1 by blocking polo-box domain-dependent protein-protein interactions. Chem Biol, 2008, 15(5):459-466.
[24] Owicki JC. Fluorescence polarization and anisotropy in high throughput screening: perspectives and primer. J Biomol Screen, 2000, 5(5):297-306.
[25] Jameson DM, Ross JA. Fluorescence polarization/anisotropy in diagnostics and imaging. Chem Rev, 2010, 110(5):2685-2708.
[26] Jameson DM, Croney JC. Fluorescence polarization: past, present and future. Comb Chem High Throughput Screen, 2003, 6(3):167-173.
[27] Golsteyn RM, Schultz SJ, Bartek J, et al. Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci, 1994, 107(Pt 6):1509-1517.
[28] Holtrich U, Wolf G, Bräuninger A, et al. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proc Natl Acad Sci U S A, 1994, 91(5):1736-1740.
[29] Rödel F, Keppner S, Capalbo G, et al. Polo-like kinase 1 as predictive marker and therapeutic target for radiotherapy in rectal cancer. Am J Pathol, 2010, 177(2):918-929.
[30] Reindl W, Yuan J, Krämer A, et al. A pan-specific inhibitor of the polo-box domains of polo-like kinases arrests cancer cells in mitosis. Chembiochem, 2009, 10(7):1145-1148.
[31] Reinson T, Henno L, Toots M, et al. The cell cycle timing of human papillomavirus DNA replication. PLoS One, 2015, 10(7):e0131675.
[32] Emmitte KA, Adjabeng GM, Andrews CW, et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubilityand reduced protein binding. Bioorg Med Chem Lett, 2009, 19(6): 1694-1697.
Author Affiliations:College of Pharmacy, Heilongjiang University of Chinese Medicine, Harbin 150040, China (ZONG Ke-xin,FAN Zhuo-wen); National Laboratory for Screening New (Microbial) Drugs, Institute of Medicinal Biotechnology, Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, China (ZONG Ke-xin, ZHANG Jing, CHEN Yun-yu, SI Shu-yi); Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China (CHEN Yun-yu)
www.cmbp.net.cnChin Med Biotechnol, 2016, 11(1):13-20
·协会之窗·
[33] Murugan RN, Park JE, Lim D, et al. Development of cyclic peptomer inhibitors targeting the polo-box domain of polo-like kinase 1. Bioorg Med Chem, 2013, 21(9):2623-2634.
Screening and anti-tumor activity of PLK1 PBD targeted inhibitors
ZONG Ke-xin, ZHANG Jing, CHEN Yun-yu, FAN Zhuo-wen, SI Shu-yi
【Abstract】
ObjectiveIn order to find lead compound which is capable, at least in vitro, of inhibiting tumor cells' growth by restraining PLK1 PBD's activity.
MethodsWe used fluorescence polarization model to screen positive compounds targeting PLK1 PBD. MTT assay was utilized to find the most sensitive cell lines of 1097C11. Effects of the positive compound on cell cycle and apoptosis were all tested by flow cytometry (FCM). The combination of compound with PLK1 PBD was determined by molecular docking. Compound's effect on cell movement was determined by cell migration experiment.
ResultsThe hit 1097C11 was found from 20 000 compounds. It could suppress the proliferation of HCT-116 cells. 1097C11 induced apoptosis, with apoptosis ratio reaching as high as 77.02% at 25 μmol/L concentration, and also led to G1/M phase arrest. Molecular docking showed that 1097C11 had good affinity with the PBD1 domain. Cell migration results showed that 1097C11 inhibited the migration of HCT-116 cells and the migration rate was as low as 33.4% in 20 μmol/L.
Conclusion1097C11 has good antitumor activity and is expected to be a lead compound targeting PLK1 relevant tumor.
【Key words】Fluorescence polarization;Antineoplastic agents;Polo-like kinase 1inhibitor
Corresponding Authors: FAN Zhuo-wen, Email:1282776014@qq.com; SI Shu-yi, Email: sisyimb@hotmail.com
收稿日期:2015-10-12
通信作者:范卓文,Email:1282776014@qq.com;司书毅,Email:sisyimb@hotmail.com
基金项目:国家自然科学基金面上项目(81370087)
DOI:10.3969/j.issn.1673-713X.2016.01.004
