负载型金属催化剂的热稳定机制
2016-03-19杨晓丽苏雄杨小峰黄延强王爱琴张涛
杨晓丽,苏雄,杨小峰,黄延强,王爱琴,张涛
(中国科学院大连化学物理研究所,辽宁 大连 116023)
负载型金属催化剂的热稳定机制
杨晓丽,苏雄,杨小峰,黄延强,王爱琴,张涛
(中国科学院大连化学物理研究所,辽宁 大连 116023)
摘要:负载型金属催化剂是一类重要的催化材料,在石油炼制、环境保护以及材料合成等领域起着重要的作用。然而,由于活性金属在反应环境下容易烧结团聚,以致活性降低乃至失活,因此,如何提高其热稳定性成为负载型金属催化剂研究的一个关键问题。概述了催化剂的金属团聚成因及其稳定机制。简要介绍了Ostwald效应以及颗粒合并长大两种团聚模型,从热力学角度解释了导致催化剂烧结团聚的原因。总结了现阶段几种提高负载型金属催化剂热稳定性能的方法,具体包括以包覆封装隔离为原理的物理方法,以及以形成化学键为基础的化学方法,可为进一步开发高热稳定性的负载型金属催化剂提供借鉴。
关键词:负载型金属催化剂;稳定性;团聚;烧结;稳定机制;稳定策略
2015-10-14收到初稿,2015-11-26收到修改稿。
联系人:张涛。第一作者:杨晓丽(1992—),女,硕士研究生。
Received date: 2015-10-14.
引 言
催化剂作为化学工业的基础,在现代工业生产活动中起着关键作用,其中90%以上的化学工业过程都是在催化剂作用的条件下实现的[1]。而负载型金属催化剂因其具有制备过程简单、高活性和选择性、低腐蚀性、易重复利用等优点,成为应用最为广泛的催化剂之一[2-4]。
负载型金属催化剂是将具有催化活性的金属组分负载在高比表面积的载体上,其催化性能和组成与结构有着直接的关系[5]。研究表明:减小金属颗粒尺寸至纳米级甚至是单原子时[6-8],不仅可以提高金属的原子利用率,而且可以利用材料的尺寸效应,表、界面效应等结构特点,提高负载型金属催化剂的反应活性[9-10]。但是随着金属颗粒尺寸的减小,其表面能剧增,金属颗粒易团聚。因此,在条件苛刻的高温高压反应过程中(如推进剂催化分解反应[11]),如何保持催化剂的高分散度以维持其催化活性仍是催化领域亟需解决的难题,这一问题也成为制约其广泛应用的瓶颈。因此制备兼具高活性和高热稳定性能的负载型金属催化剂仍是工业催化领域重要的研究方向。
随着现代表征技术手段的日益进步,对催化剂的热稳定性与其结构本身和活性金属所处环境等因素之间关系的认识也日渐明朗,并且对催化剂在反应过程中活性金属团聚过程的观测也逐渐清晰[12-13]。据此,研究人员依据对活性金属稳定作用的理解,设计了不同结构类型的负载型金属催化剂,提出一系列提高其热稳定性能的方法,根据其原理大致可以分为物理方法和化学方法。本文重点对负载型金属催化剂团聚的机理分析、稳定策略、优化的催化剂制备方法及其在催化反应中的应用等方面进行了综述。
1 团聚诱因分析
在实际的反应环境中,负载型金属催化剂的金属颗粒会受到自身以及周围物理化学条件的影响,离开载体表面原位点,发生表面扩散和表面聚集并烧结[14]。具体来看,可以将引起烧结团聚的原因分为物理因素和化学因素的诱导。
物理因素主要是热诱导。当环境温度达到0.3Tm(Tm为催化剂金属颗粒的泰曼温度)时,金属颗粒发生晶格表面质点的迁移;当温度为0.5Tm时,便发生晶格体相内的迁移[1]。因此,在高温环境中金属颗粒的动能增加,更容易发生迁移的行为,金属的表面扩散速率会增大,导致颗粒之间相互碰撞的概率增加,发生团聚现象。
化学因素的诱导是多方面的,也是复杂的。当反应物吸附在金属颗粒表面时,会与金属原子之间形成化学键,弱化金属原子间相互作用力,促使金属原子的迁移扩散,加快催化剂的失活[1]。此外,环境的气氛也会对负载型金属催化剂的稳定性能产生影响。一般情况下,在氧气气氛中,由于金属氧化物易沿着表面发生移动,导致金属的烧结速度相对于氢气或惰性环境中显著增加,如Pt族催化剂在氧气氛围中比在惰性氛围中原子移动速度快得多[15]。
2 团聚方式分析
负载型金属催化剂暴露在反应环境中时,受周围物理化学因素的影响,在Gibbs自由能的驱动下小的颗粒会发生表面扩散形成较大的三维颗粒。根据粒子的运动方式,可以将粒子的生长机理概括为Ostwald效应和粒子迁移长大两种模型[16],如图1所示。在不同的反应阶段,由不同的机理引起粒子发生变化,对负载型金属催化剂的团聚起主要作用。
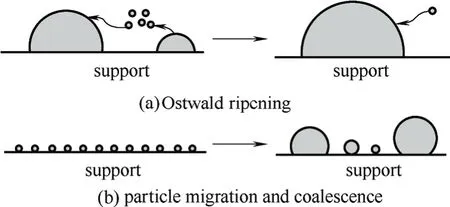
图1 负载型金属催化剂的团聚模型Fig.1 Aggregation model of supported metal catalyst
2.1 Ostwald效应
Ostwald效应是指在一定条件下,由于小颗粒化学势比较高,原子从小颗粒逐渐分离并沉积到大颗粒上的一种热力学行为[17-19]。Hansen等[16]提出,当颗粒很小的时候,它是导致催化剂失活的主要原因。
对于负载型金属催化剂,随着金属颗粒尺寸的减小,更容易发生Ostwald熟化现象。Li等[20]对负载型金属催化剂上存在的Ostwald效应进行理论计算分析,从热力学的角度给予解释:小颗粒由于自身的化学势能比较高,热稳定性能低,易发生团聚。Ostwald效应的熟化能应包含粒子的扩散能和离开金属颗粒表面或黏附在另外的颗粒表面的界面能两部分,催化剂的组成以及周围的环境对熟化能会产生相应的影响。
反应物的存在会在一定程度上促进Ostwald效应的发生。一方面,反应物会吸附在金属颗粒表面,放出一部分能量,使总的熟化能降低,有助于发生熟化现象[20]。另一方面,金属与反应物形成的过渡物种也会增加颗粒的迁移扩散速率,促进Ostwald熟化现象的发生[15]。如被广泛用于工业甲醇合成和低温水煤气变换反应的Cu/ZnO/Al2O3催化剂,当存在CO时,Cu粒子通过形成Cu-CO物种可增加其在ZnO表面的迁移速率,促进熟化现象的发生,从而加速催化剂的失活[21]。
根据发生Ostwald效应的原理分析,可以通过选择与金属原子具有高界面能的载体或者制备高规整均一性好的催化剂,从而抑制金属颗粒的熟化速率,提高负载型金属催化剂的热稳定性能[20]。
2.2 粒子迁移长大
粒子迁移长大是单个晶粒沿着载体表面移动做类布朗运动,持续迁移扩散,并由粒子相互聚集而合并生长的现象[22]。如Ir单原子负载在Y型分子筛上,会从单原子经过B + B → C生成较小的团簇,再经过B + C → 1.5C反复循环,慢慢合并为大的团簇[23]。它包含粒子的迁移和聚集两个步骤,其中聚集过程又包括两颗粒相连结处的生长和两个相互结合的颗粒在外形上逐渐变成具有低能量的排列方式两个过程。研究表明,对催化有意义的体系一般来说其限制步骤是颗粒移动阶段而不是颗粒聚集阶段[15]。
另外,粒子迁移长大是一个自催化的过程。粒子间通过化学键合作用合并长大并放出热量,该热量可促使粒子的进一步迁移长大。Shirakawa等[24]对Au/SiO2催化剂的团聚现象进行研究,发现刚开始时Au的迁移合并速率较慢,但随着时间的增长,Au颗粒的迁移长大速度在自催化效应的影响下逐渐增加。
在高温环境中,以上两种方式共同作用引起了负载型金属催化剂的烧结团聚。为了降低因金属颗粒的烧结团聚而引起的催化剂失活程度,研究者研究设计了不同的方案,以下针对不同的稳定策略分别进行概述。
3 稳定策略
由前文提到的金属颗粒发生的Ostwald效应和颗粒迁移长大两种团聚方式分析,金属颗粒的团聚主要由迁移扩散和附着聚积两步组成[25],据此,可以从这两方面入手来提高负载型金属催化剂的热稳定性能。一是在空间上隔离金属颗粒防止其相互聚集长大,二是通过增强金属在载体上的附着强度来降低金属颗粒的迁移扩散能力。根据提高催化剂热稳定性能的原理分析,可以将稳定策略分为物理方法和化学方法。
3.1 物理方法
基于物理方法的稳定策略是将负载型金属催化剂的金属颗粒进行包封分装,从而使其在各自限定的空间内运动,颗粒间相互隔离,提高发生团聚的能垒从而达到稳定的效果。采用该原理稳定负载型金属催化剂的方法有很多种:20世纪80年代,人们发现核壳结构可以将金属颗粒包覆,使之相互隔离[26];随后又发现具有孔道结构的载体可以将金属颗粒的运动限制在一定范围内,减小颗粒团聚的概率[27];最近几年,人们利用原子层沉积的技术在催化剂表面涂覆上保护层,以减缓金属颗粒的迁移扩散速率[28]。
3.1.1 核壳结构 核壳结构是以一个尺寸在微米至纳米级别的颗粒为核,在其表面包覆上一层或者数层均匀的纳米壳层而形成的一种复合多相结构,核和壳之间以物理或化学作用相互连接[29]。壳可以将纳米颗粒分别包封,使颗粒之间相互隔离,从而防止颗粒之间的迁移聚积,提高负载型金属催化剂的抗烧结能力,如图2所示。核壳结构是最常用的提高负载型金属催化剂稳定性能的方法之一[30-31]。

图2 核壳结构Fig.2 Core-shell structure
传统的核壳结构是采用自组装的方法,在纳米颗粒的表面包裹上一层外壳。其中,外壳可作为载体包裹在金属颗粒的表面,或者也可以直接包裹在负载型金属催化剂的球形颗粒上[32-33]。Joo等[34]在Pt纳米颗粒外包裹上一层介孔的SiO2外壳,形成Pt@mSiO2结构。研究发现,设计合成的Pt@mSiO2催化剂在750℃下焙烧后仍能保持原来的核壳结构,如图3所示。并且该催化剂对乙烯加氢和CO氧化反应的催化性能可以长时间保持稳定,具有很好的抗烧结能力。Lee等[35]在易团聚失活的Pt/SiO2催化剂上包覆上介孔的SiO2层,形成Pt/SiO2@SiO2的核壳结构来提升催化剂的稳定性。他们将裸露的催化剂和形成核壳结构的催化剂同时焙烧,发现未被保护的催化剂在600℃时发生明显的烧结;而经过包覆保护的催化剂在800℃的高温下,依然可以保持稳定,未发生明显的团聚现象。
随着纳米技术手段的进步,在传统的核壳结构基础上又出现了其他新颖的核壳结构,如核-多壳和多核-壳结构[36]。Schwarze等[37]采用亲水的超支化聚甘油为核心,疏水的十八烷基为内层,亲水的聚乙二醇为外层,合成了Pt@CMS与Pd@CMS的核-壳-壳结构。Chen等[38]利用包裹10 nm Pd的Pd@SiO2小球与Ce(NO3)3在乙二醇-水-乙酸混合液中反应,制备了含多个Pd@SiO2核的Pd@SiO2@m-CeO2催化剂。研究发现,上述两种催化剂均具有较强的热稳定性能,在长时间的高温反应后核壳结构没有明显的变化。将其进行多次循环使用,活性依然可以保持在一个稳定的状态。

图3 Pt@mSiO2焙烧后的TEM图[34]Fig.3 TEM images of Pt@mSiO2after calcination[34]
核壳结构在空间上使金属颗粒间相互隔离,有助于防止颗粒发生团聚现象。但该结构也具有一定的局限性,有效的核壳结构应该是允许反应物分子从外面通过壳层到达金属颗粒表面的,即壳层应有一定的通道使反应发生[39]。因此,如何制备出具有均一大小孔尺寸的壳结构是该方法的主要突破点。
3.1.2 孔道限域作用 孔道限域作用是使用具有一定孔道或笼状结构的材料封装金属颗粒,利用其空间结构来限制金属颗粒的迁移扩散行为,从而阻止颗粒间发生团聚,并提供分子反应通道和空间。按照载体的孔道尺寸大小可将其分为微孔和介孔材料,如图4所示。
分子筛拥有规整均一的孔道分布和笼结构,是常用的微孔限域材料。早在20世纪90年代,Rao 等[42]发现Rh团簇可以很好地分散稳定在NaY分子筛的超笼中。后来,Lu等[41]也发现高规整的NaY分子筛可以稳定贵金属Au,他们利用球差校正扫描透射电子显微镜观测到Au复合物呈单原子状态稳定分散在NaY分子筛上。并进一步将该催化剂用于CO氧化反应,反应后未观测到Au团簇的存在,结果表明由于分子筛的孔道限域作用以及孔笼内碱金属离子形成的静电场作用,Au原子被稳定在NaY分子筛中。除了Y型分子筛,其他类型的分子筛也都有着类似的作用。Laursen等[43]采用水热法合成了全硅沸石silicalite-1包封着Au纳米颗粒的催化剂。研究发现,该催化剂在550℃的环境中焙烧后,Au纳米颗粒的尺寸基本保持不变。继续将该催化剂在500℃的O2气氛围中焙烧,包封在分子筛的孔道或腔中的小颗粒仍未发生团聚现象。Wu等[44]采用水热合成方法将贵金属Pt、Pd、Rh、Ir、Re和 Ag封装在LTA分子筛中合成M/LTA催化剂。将其在O2氛中处理,在300~600℃的温度范围内金属团簇都可以维持原有的尺寸。在LTA笼结构的限域下,该催化剂具有良好抗高温烧结能力,并且对于反应物的进入具有选择性,使催化剂具有一定的抵抗毒性物质的能力。

图4 孔道结构的限域作用[40-41]Fig.4 Confined effect of porous structure[40-41]
虽然分子筛有着高度规整均一的孔笼结构,但其孔道尺寸过小,一部分分子筛对于金属前体的引入有所限制,无法负载大的金属团簇,并且催化剂对存在较大分子的原料或产物的催化反应具有局限性[45],所以利用介孔材料的限域作用也是十分必要的[40,46]。Jiang等[47]将Pd纳米颗粒封入SBA-15孔道中,合成的Pd/SBA-15催化剂不仅对烯丙醇有着高加氢活性,而且多次循环使用后并未发生团聚现象。在此基础上,将该催化剂在常温下放置一个月后再次检测其催化性能,发现活性并未降低。Bao 等[48]同样利用SBA-15的孔道限域作用封装Ag颗粒。实验表明,以上介孔SiO2材料限域的金属颗粒均有较高的抗烧结能力。此外,介孔碳材料以及介孔Al2O3材料也可以用来包封限域金属纳米颗粒。Ribeiro等[49]用介孔Al2O3包封Pt纳米颗粒,并将其进行高温热处理,实验证明该催化剂具有优良的抗烧结能力。为进一步提高催化剂的抗高温烧结能力,Zhang等[50]采用“一锅法”将Ir颗粒镶嵌在有序介孔碳材料的孔壁上,经800℃高温处理后Ir的粒径在2 nm左右,在肼分解反应中表现出了高的活性和热稳定性。
3.1.3 原子层沉积法 作为一种表面控制的、自限制性的化学气相沉积方法,原子层沉积技术(ALD)通过控制活性金属在载体上的沉积循环次数和处理温度可以实现对金属颗粒大小的调控[51-53]。Lu等[54]通过这种方法实现了1~3 nm的Pd颗粒在Al2O3和TiO2载体上的生长。对于已制备成的负载型催化剂,ALD技术还能在颗粒表面沉积上保护层,将氧化物涂层可控沉积在基质或颗粒的表面,给颗粒的迁移扩散设置一个物理障碍,降低其迁移扩散速率从而提高负载型金属催化剂的稳定性能,如图5所示。
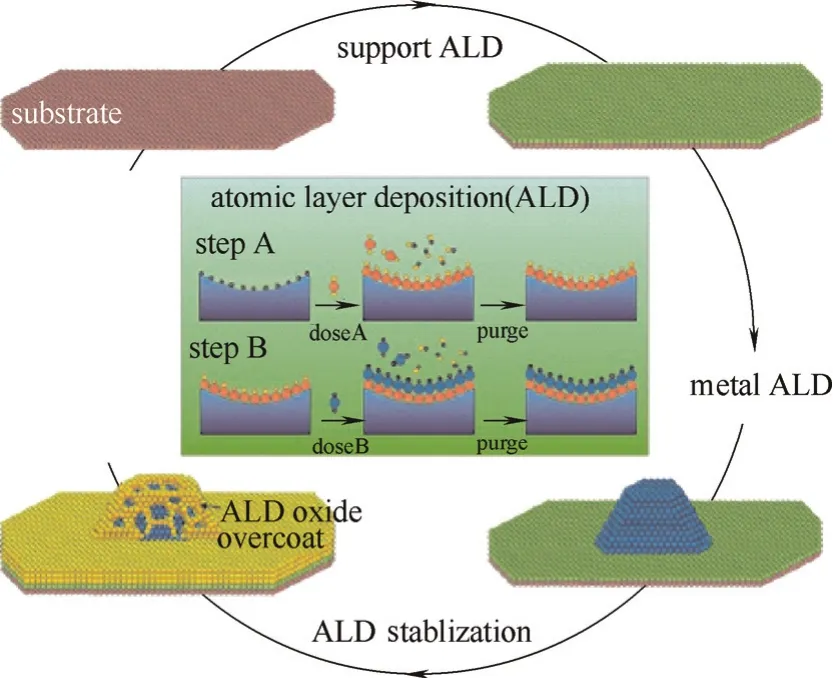
图5 ALD作用原理[55]Fig.5 Principle diagram of ALD technique[55]
ALD技术可以选择性地包封金属颗粒、载体或催化剂,既不损失负载型金属催化剂的活性,又能提高其热稳定性能[55]。Lu等[56]采用ALD技术将Al2O3沉积在Pd/Al2O3的表面,不仅避免了Pd催化剂的烧结,而且有效抑制了反应积炭,从而增加了催化剂的稳定性能。Liang等[57]在Pt/SiO2的表面沉积上Al2O3涂层,提高了催化剂的稳定性能,使其在800℃的温度下焙烧,依然可以保持颗粒状而不发生烧结团聚。Sung等[58]在Ag纳米颗粒表面通过ALD技术涂覆上一层Al2O3,然后比较了裸露的Ag纳米颗粒和Al2O3-ALD-Ag催化剂对飞秒激光脉冲的稳定性。通过紫外可见吸收光谱观测两者的结构变化,结果表明Al2O3-ALD-Ag的稳定性比Ag纳米颗粒提高了10倍。解释为由于ALD涂层,使表面Ag原子密度降低,导致表面熔点升高,增加了颗粒的稳定性能。以上结果均表明,ALD 技术可以有效地提高催化剂的稳定性能。
ALD技术最大的优点就是可以精确控制涂层的厚度。但受技术规模和经济的制约,采用该方法来制备催化剂距离工业化还有一定的路程。
3.2 化学方法
化学方法稳定负载型金属催化剂,一般是依靠化学键合作用来提高金属和载体的附着能,从而减缓金属颗粒的迁移扩散,阻止颗粒的烧结团聚,提高催化剂的稳定性能。通过选择合适的载体与金属颗粒形成共价键,或者加入另外一种金属形成金属键,是提高催化剂热稳定性能的有效方法。
3.2.1 共价键合作用 负载型金属催化剂的载体一般选用金属氧化物、分子筛或是碳材料,来提供大的比表面积分散担载活性金属。近年来研究发现,载体还会与金属颗粒形成强的共价键合作用,并发生电子转移,从而提高催化剂的稳定性能[59-61]。选择合适的载体,增加与金属颗粒之间相互作用力,可以显著提高负载型金属催化剂稳定性能[62]。
(1)非还原性载体 非还原性载体不含有可变化学价金属元素,主要有Al2O3、SiO2以及分子筛等。
金属颗粒与载体形成的键合作用可以将金属原子固定在相应的位置,提高催化剂的稳定性能。概括来讲,金属主要通过O原子与载体发生化学键合,形成稳定的共价键合作用[7]。Kwak等[63]利用魔角旋转核磁共振光谱结合其他的表征手段检测到Pt负载在γ-Al2O3载体上的锚定位点,结果显示Pt的引入使五配位的Al转变为六配位的Al,即Pt通过O与Al发生键合作用,使配位未饱和的Al达到配位饱和,此键合作用是Pt/γ-Al2O3具有高稳定性能的原因。催化剂的稳定性会随着金属与O原子间键合作用的增强而增强。Yang等[64]发现Au负载在KLTL或者MCM-41的孔道内时,添加碱基离子会使单核Au变得稳定。研究发现,碱基离子的存在使惰性载体上产生单核的Au-O(OH)x物种,分散性提高。在此基础上,作者结合理论计算,证实通过改变AuOxNa9团簇中氧原子的数量可以调节Au与O之间的键合作用,O原子数量越多,Au携带电荷越大,即Au与O的共价键合作用越强,催化剂的稳定性能也越高,如图6所示。Li等[65]利用DFT计算和实验测试得到贵金属Rh、Ir、Pt的晶面可以和MgAl2O4尖晶石的(111)晶面相互匹配,与其表面的O原子形成一定的键合作用。研究发现,该催化剂在800℃的高温下氧化老化处理1周,金属颗粒的尺寸仍可以保持在1~3 nm,具有很好的抗烧结能力。
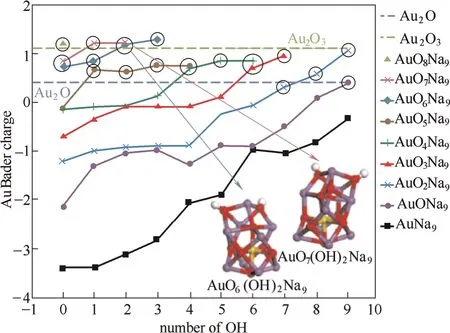
图6 AuOx(OH)yNa9团簇中Au的电荷变化[64]Fig.6 Au Bader charge in AuOx(OH)yNa9cluster[64]
金属除了可以与O原子相连形成键合作用外,还可以与载体表面其他原子相连形成共价键合作用。如Bao等[66]通过对硅酸亚铁和二氧化硅共同熔化,用HNO3沥滤、干燥获得了 Fe©SiO2催化剂。将该催化剂应用于甲烷制乙烯、芳香烃的反应中,研究发现在1090℃的高温下反应60 h后,活性未发生明显的降低。通过HAADF-STEM观察催化剂的形貌,发现铁原子以单点的形式分布在SiO2基质上;用X射线吸收近边结构(XANES)进一步验证了反应过程中Fe镶嵌在Si和C之间形成了键合作用,保证该催化剂高温下的稳定。
(2)可还原性载体 若载体中含有可变价金属元素,则为可还原性载体,如CeO2、Fe2O3、TiO2、MnOx等。在还原过程中,金属元素化学价降低,产生氧空位。金属颗粒会通过载体表面的氧空穴与其形成共价键合作用,促进负载型金属催化剂热稳定性能的提高[67]。
相对于非还原性载体,金属通过氧空位与可还原性载体产生的结合能更大。Farmer等[68]研究当Ag负载在不同的载体上时,如CeO2和MgO,产生的吸附能大小不同,并发现利用可还原性CeO2-x作为载体时,负载的Ag有着更强的抗烧结能力。相比于MgO载体,CeO2-x载体具有大量的氧空位,银离子更易通过氧空位键合在载体上(比其他位置稳定性更强),形成三维岛状结构。研究还发现Ag纳米粒子的稳定性能会随着氧空位的增多而增强。Shen等[69]也研究发现2~4 nm的Au团簇通过氧空位与CeO2载体形成强的键合作用,在氧化还原催化循环的过程仅发现了Au颗粒形状微弱的变化。Zhang等[8]采用改进的共沉淀方法制备了单原子催化剂Pt1/FeOx,并结合X射线吸收精细结构谱(XAFS)和傅里叶变换红外CO吸附研究表明Pt和FeOx之间形成了Pt-O-Fe键,促使Pt以单原子的形式稳定存在,如图7所示。将该催化剂用于PROX反应,研究发现,催化剂运转1000 min后对CO的转化率和CO2的选择性仍没有衰退,表明该催化剂具有较好的稳定性。

图7 Pt1/FeOx催化剂[8]Fig.7 Pt1/FeOxcatalyst[8]
当活性组分和载体的晶格匹配程度高时,通过氧空位形成的键合作用更强。Si等[70]研究了不同晶面CeO2对Au稳定性能的影响,分别用棒状、立方状和多面体状的CeO2来负载Au。研究发现:在纳米立方的{100}晶面上,键合作用很弱;但在纳米棒状的{110}晶面上,键合作用要强得多。虽然在对催化剂的表征和反应性能评价中平均每个原子的活性差不多,但金属颗粒的稳定性却表现出明显的差异。Huang等[71]选用与RuO2同为金红石晶型的TiO2为载体,利用其晶型相同、晶格匹配程度高的特点,获得了具有高热稳定性的Ru/TiO2催化剂,在CO2甲烷化以及高浓度N2O分解反应中表现出了高的活性以及热稳定性。
相比于其他方法,采用金属颗粒和载体形成强的共价键合作用来提高负载型金属催化剂稳定性的策略更具有普遍适用性,因而被广泛应用于催化剂热稳定性能的提升。
3.2.2 金属键合作用 在高温环境中,单一金属容易发生烧结团聚的现象,可以通过在负载型金属催化剂中添加另一种金属组分,通过形成合金,依据合金中不同元素组分比单一金属更强的金属-金属键合作用,从而调变金属的物理和化学性质,促使其结构更加致密,热稳定性增强,提高其抗烧结能力[72-73]。
众所周之,Au基催化剂在多个反应表现出优异的低温催化活性,但易团聚,通过引入第二金属组分Pd形成AuPd合金,甚至是更低熔点的Cu或Ag等,均能够显著提升其热力学稳定性[74]。如Zhang 等[75]将Au原子固定在Pd团簇的端位,形成类似“皇冠上的珍珠”状的结构,其原理就是利用合金化来稳定Au原子;Zhang等[76]通过调控Au:Pd比例,在Au纳米颗粒上实现了Pd的单原子分散,制备的Au4Pd/树脂催化剂在乌尔曼反应中重复使用8次活性不降低,表现出了优异的稳定性。同时,Cheng 等[77]发现在Au55团簇中掺杂一个Cu原子,形成了Cu核Au壳的团簇,能够大幅度提高其熔点。Liu 等[78]也对Ag与Au形成的一系列Au38-xAgx(x=0, 1, 2)合金进行了DFT计算,研究发现当Ag沉积在Au38团簇表面上后会扩散到团簇内部,形成Au37Ag团簇,相应的平均结合能增加,即熔点升高,如图8所示;并发现其熔点随着Ag含量的增加而逐渐升高,其稳定性能也逐渐提高,说明合金熔点的升高是其稳定性提高的重要原因。与金的合金作用相似,Zhang等[79-80]还通过在Ni基催化剂中加入另外一种金属如Pt、Ir等,通过形成Ni基合金,不仅提高了其抗烧结能力,而且显著改变了其肼分解制氢活性和选择性;Cao等[81]在低熔点的Pt催化剂中加入高熔点的金属Rh,形成PtRh合金并负载在六铝酸盐载体上,经850℃焙烧后金属颗粒尺寸未发生明显变化,表现出了优异的抗烧结能力和热稳定性能。
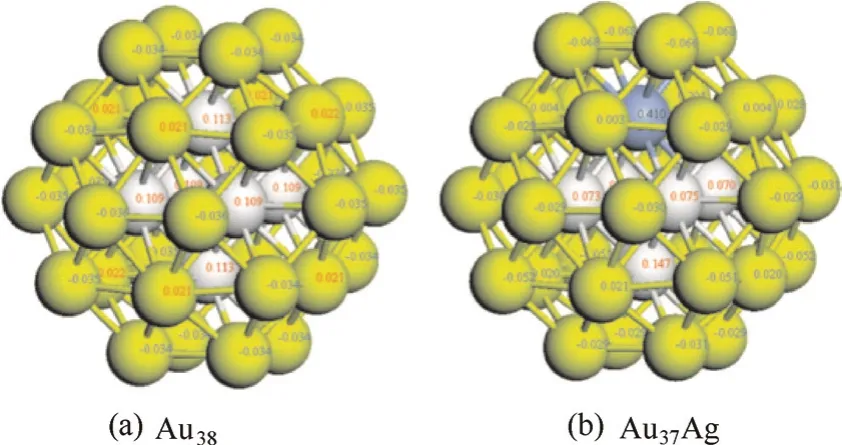
图8 团簇电荷分布[78]Fig.8 Charge distribution of clusters[78]yellow: Au(inside);gray: Au(outside); blue: Ag
此外,合金结构在反应条件下发生分相,其中一种组分变成氧化物,成为稳定活性的金属的氧化物壳层,形成M1@M2O结构。这样,第二金属起到类似于“纳米胶”的作用,从而能够显著提高母体金属的热稳定性。如在惰性载体负载的Au-Ag和Au-Cu合金催化剂上,研究者发现Ag或Cu在氧化气氛下在Au粒子及载体接触表面可形成氧化物壳层(Ag2O或CuO),从而抑制Au颗粒的迁移,提高Au颗粒的抗高温烧结能力[82]。
综上,加入另外一种金属元素形成合金催化剂,可以提高金属的熔点或者在反应条件下形成M1@M2O结构,从而提高负载型金属催化剂的抗烧结能力和催化性能。
4 展 望
提高催化剂的热稳定性一直是催化领域研究的热点和难点。本文综述了目前在提高催化剂热稳定性能的最新探索,主要包括以包覆封装隔离为原理的物理方法以及以形成化学键为基础的化学方法。
但是,催化剂热稳定性能的提升依然存在很大的挑战。首先,许多提高催化剂稳定性能的方法都是以损失活性的代价来实现的,这样对于贵金属而言经济性反而下降。其次,很多方法只能针对某些特定的催化反应体系,缺乏必要的普适性。尽管如此,随着表征技术的提高和理论计算的发展,以及对催化剂的稳定性能与结构认识的深入,必将能够实现高热稳定性能的负载型金属催化剂的理性设计,进而为工业催化剂的生产提供重要的借鉴。
References
[1] 黄仲涛. 工业催化剂手册[M]. 北京: 化学工业出版社, 2004. HUANG Z T. Handbook of Industrial Catalysts[M]. Beijing: Chemical Industry Press, 2004.
[2] 曾成华. 负载型金属催化剂的研究进展 [J]. 攀枝花学院学报, 2006, 23(2): 110-114. DOI: 10.3969/j.issn.1672-0563.2006.02.033. ZENG C H. The research progress of supported metal catalysts [J]. Journal of Panzhihua University, 2006, 23(2): 110-114. DOI: 10.3969/j.issn.1672-0563.2006.02.033.
[3] 郑双双, 刘利平. 负载型金属催化剂制备新技术研究进展 [J]. 广东化工, 2012, 39(9): 12-13. DOI: 10.3969/j.issn.1007-1865. 2012.09.006. ZHENG S S, LIU L P. Research progress of new technology on supported metal catalyst preparation [J]. Guangdong Chemical Industry, 2012, 39(9): 12-13. DOI: 10.3969/j.issn.1007-1865. 2012.09.006.
[4] GATES B C. Supported metal cluster catalysts: progress and perspectives// Abstracts of Papers of the American Chemical Society[C]. 2001: 222, U198.
[5] 张以敏, 姜浩锡. 超临界流体沉积技术制备负载型金属催化剂的研究进展 [J]. 化工进展, 2013, (8): 1825-1831. DOI: 10.3969/j.issn. 1000-6613.2013.08.018. ZHANG Y M, JIANG H X. Preparation of supported metal catalyst via supercritical fluid deposition [J]. Chemical Industry and Engineering Progress, 2013, (8): 1825-1831. DOI: 10.3969/j.issn.1000-6613.2013.08.018.
[6] THOMAS J M, JOHNSON B F G, RAJA R, et al. High-performance nanocatalysts for single-step hydrogenations [J]. Cheminform, 2003, 36(16): 20-30.
[7] YANG X F, WANG A Q, QIAO B T, et al. Single-atom catalysts: a new frontier in heterogeneous catalysis [J]. Accounts of Chemical Research, 2013, 46(8): 1740-1748.
[8] QIAO B T, WANG A Q, YANG X F, et al. Single-atom catalysis of CO oxidation using Pt1/FeOx[J]. Nature Chemistry, 2011, 3(8): 634-641.
[9] JAIN P K, HUANG X, EL-SAYED I H, et al. Noble metals on the nanoscale: optical and photothermal properties and some applications in imaging, sensing, biology, and medicine [J]. Accounts of Chemical Research, 2008, 41(12): 1578-1586.
[10] TOH H S, COMPTON R G. ‘Nano-impacts’: an electrochemical technique for nanoparticle sizing in optically opaque solutions [J]. Chemistryopen, 2015, 4(3): 261-263.
[11] 李令成, 蓝蕴基. 肼分解催化剂进展 [J]. 工业催化,1994, 1(1): 3-7. LI L C, LAN Y J. Process in hydrazine decomposition catalysts [J]. Industrial Catalysis, 1994, 1(1): 3-7.
[12] 吴霞. 负载金属催化剂的结构性能表征 [J]. 广东化工, 2012, 39(9): 39. DOI: 10.3969/j.issn.1007-1865.2012.09.020. WU X. Structure characterization of supported metal clusters [J]. Guangdong Chemical Industry, 2012, 39(9): 39. DOI: 10.3969/j.issn. 1007-1865.2012.09.020.
[13] 杨春雁, 杨卫亚, 凌凤香, 等. 负载型金属催化剂表面金属分散度的测定 [J]. 化工进展, 2010, 29(8): 1468-1473. YANG C Y, YANG W Y, LING F X, et al. Determination of metal dispersion on supported metal catalyst surface [J]. Chemical Industry and Engineering Progress, 2010, 29(8): 1468-1473.
[14] LORIA H, PEREIRA-ALMAO P, SCOTT C E. Determination of agglomeration kinetics in nanoparticle dispersions [J]. Industrial & Engineering Chemistry Research, 2011, 50: 8529-8535.
[15] 萨特菲尔德. 实用多相催化[M]. 庞礼, 译. 北京: 北京大学出版社,1990. SATTERFIELD C N. Practical Heterogeneous Catalysis[M]. PANG L, trans. Beijing: Beijing University Press, 1990.
[16] HANSEN T W, DELARIVA A T, CHALLA S R, et al. Sintering of catalytic nanoparticles: particle migration or Ostwald ripening? [J]. Accounts of Chemical Research, 2013, 46(8): 1720-1730.
[17] VOORHEES P W. The theory of Ostwald ripening [J]. Journal of Statistical Physics, 1985, 38(1/2): 231-252.
[18] KISTAMURTHY D, SAIB A M, MOODLEY D J, et al. Ostwald ripening on a planar Co/SiO2catalyst exposed to model Fischer-Tropsch synthesis conditions [J]. Journal of Catalysis, 2015, 328: 123-129.
[19] LIU B, ZENG H C. Symmetric and asymmetric Ostwald ripening in the fabrication of homogeneous core-shell semiconductors [J]. Small, 2005, 1(5): 566-571.
[20] OUYANG R H, LIU J X, LI W X. Atomistic theory of Ostwald ripening and disintegration of supported metal particles under reaction conditions [J]. Journal of the American Chemical Society, 2013, 135(5): 1760-1771.
[21] RASMUSSEN D B, JANSSENS T V W, TEMEL B, et al. The energies of formation and mobilities of Cu surface species on Cu and ZnO in methanol and water gas shift atmospheres studied by DFT [J]. Journal of Catalysis, 2012, 293(1): 205-214.
[22] FINNEY E E, SHIELDS S P, BUHRO W E, et al. Gold nanocluster agglomeration kinetic studies: evidence for parallel bimolecular plus autocatalytic agglomeration pathways as a mechanism-based alternative to an avrami-based analysis [J]. Chemistry of Materials, 2012, 24(10): 1718-1725.
[23] BAYRAM E, LU J, AYDIN C, et al. Agglomerative sintering of an atomically dispersed Ir1/zeolite Y catalyst: compelling evidence against Ostwald ripening but for bimolecular and autocatalytic agglomeration catalyst sintering steps [J]. ACS Catalysis, 2015, 5(6): 3514-3527.
[24] SHIRAKAWA H, KOMIYAMA H. Migration-coalescence of nanoparticles during deposition of Au, Ag, Cu, and GaAs on amorphous SiO2[J]. Journal of Nanoparticle Research, 1999, 1(1): 17-30.
[25] ZHANG W J, MISER D E. Coalescence of oxide nanoparticles: in situ HRTEM observation [J]. Journal of Nanoparticle Research, 2006, 8(6): 1027-1032.
[26] LUND C R F, DUMESIC J A. Strong oxide-oxide interactions in silica-supported magnetite catalysts(Ⅳ): Catalytic consequences of the interaction in water-gas shift [J]. Journal of Catalysis, 1982, 76(82): 93-100.
[27] SHEN G C, ICHIKAWA M. Methane hydrogenation and confirmation of CHxintermediate species on NaY encapsulated cobalt clusters and Co/SiO2catalysts: EXAFS, FTIR, UV characterization and catalytic performances [J]. Journal of the Chemical Society Faraday Transactions, 1997, 93(6): 1185-1193.
[28] RASHKEEV S N, DAI S, OVERBURY S H. Modification of Au/TiO2nanosystems by SiO2monolayers: toward the control of the catalyst activity and stability [J]. Journal of Physical Chemistry C, 2010, 114(7): 2996-3002.
[29] 李雷, 李彦兴, 姚瑶, 等. 核壳结构纳米材料的创制及在催化化学中的应用 [J]. 化工进展, 2013, 25(10): 1681-1690. LI L, LI Y X, YAO Y, et al. Process and prospective in fabrication and application of core-shell structure nanomaterials in catalytic chemistry [J]. Chemical Industry and Engineering Progress, 2013, 25(10): 1681-1690.
[30] LONG N V, YANG Y, THI C M, et al. The development of mixture, alloy, and core-shell nanocatalysts with nanomaterial supports for energy conversion in low-temperature fuel cells [J]. Nano Energy, 2013, 2(5): 636-676.
[31] YONG W, MIN Y L, YU S H. Synthesis of silica/carbon-encapsulated core-shell spheres: templates for other unique core-shell structures and applications in in situ loading of noble-metal nanoparticles [J]. Langmuir, 2008, 24(9): 5024-5028.
[32] HE B B, ZHAO Q G, ZENG Z G, et al. Effect of hydrothermal reaction time and calcination temperature on properties of Au@CeO2core-shell catalyst for CO oxidation at low temperature [J]. Journal of Materials Science, 2015, 50 (19): 6339-6348.
[33] ADIJANTO L, SAMPATH A, YU A S, et al. Synthesis and stability of Pd@CeO2core-shell catalyst films in solid oxide fuel cell anodes [J]. ACS Catalysis, 2013, 3(8): 1801-1809.
[34] JOO S H, PARK J Y, TSUNG C K, et al. Thermally stable Pt/mesoporous silica core-shell nanocatalysts for high-temperature reactions [J]. Nature Materials, 2009, 8(2): 126-131.
[35] LEE I, ZHANG Q, GE J P, et al. Encapsulation of supported Pt nanoparticles with mesoporous silica for increased catalyst stability [J]. Nano Research, 2011, 4(1): 115-123.
[36] LAUHON L J, GUDIKSEN M S, WANG C L, et al. Epitaxial core-shell and core-multishell nanowire heterostructures [J]. Nature, 2002, 420(6911): 57-61.
[37] SCHWARZE M, KEILITZ J, NOWAG S, et al. Quasi-homogeneous hydrogenation with platinum and palladium nanoparticles stabilized by dendritic core-multishell architectures [J]. Langmuir, 2011, 27(10): 6511-6518.
[38] CHEN C, FANG X L, WU B H, et al. A multi-yolk-shell structured nanocatalyst containing sub-10 nm Pd nanoparticles in porous CeO2[J]. Chemcatchem, 2012, 4(10): 1578-1586.
[39] ZHANG Q, LEE I, JOO J B, et al. Core-shell nanostructured catalysts [J]. Accounts of Chemical Research, 2012, 46(8): 1816-1824.
[40] TIAN H, LI X, ZENG L, et al. Recent advances on the design of group Ⅷ base-metal catalysts with encapsulated structures [J]. ACS Catalysis, 2015, 5(8): 4959-4977.
[41] LU J, AYDIN C, BROWNING N D, et al. Imaging isolated gold atom catalytic sites in zeolite NaY [J]. Angewandte Chemie International Edition, 2012, 51(24): 5842-5846.
[42] RAO L F, PRUSKI M, KING T S. Structure and stability of rhodium clusters in NaY studied by NMR and FTIR [J]. The Journal of Physical Chemistry B, 1997, 101(29): 5717-5724.
[43] LAURSEN A B, HOJHOLT K T, LUNDEGAARD L F, et al. Substrate size-selective catalysis with zeolite-encapsulated gold nanoparticles [J]. Angewandte Chemie International Edition, 2010, 122(20): 3582-3585.
[44] WU Z J, GOEL S, CHOI M, et al. Hydrothermal synthesis of LTA-encapsulated metal clusters and consequences for catalyst stability, reactivity, and selectivity [J]. Journal of Catalysis, 2014, 311: 458-468.
[45] 徐如人. 分子筛与多孔材料化学[M]. 北京: 科学出版社, 2004. XU R R. Molecular Sieve and Porous Material Chemistry [M]. Beijing: Science Press, 2004.
[46] HUANG W, KUHN J N, TSUNG C K, et al. Dendrimer templated synthesis of one nanometer Rh and Pt particles supported on mesoporous silica: catalytic activity for ethylene and pyrrole hydrogenation [J]. Nano Letters, 2008, 8(7): 2027-2034.
[47] JIANG Y J, GAO Q M. Heterogeneous hydrogenation catalyses over recyclable Pd(0) nanoparticle catalysts stabilized by PAMAM-SBA-15 organic-inorganic hybrid composites [J]. Journal of the American Chemical Society, 2006, 128(3): 716-717.
[48] ZHANG H, SUN J M, MA D, et al. Unusual mesoporous SBA-15 with parallel channels running along the short axis [J]. Journal of the American Chemical Society, 2004, 126(24): 7440-7441.
[49] RIBEIRO R U, MEIRA D M, RODELLA C B, et al. Probing the stability of Pt nanoparticles encapsulated in sol-gel Al2O3using in situ and ex situ characterization techniques [J]. Applied Catalysis A General, 2014, 485: 108-117.
[50] GAO P, WANG A, WANG X, et al. Synthesis of highly ordered Ir-containing mesoporous carbon materials by organic-organic self-assembly [J]. Chemistry of Materials, 2008, 20(5): 1881-1888.
[51] O'NEILL B J, JACKSON D H K, LEE J, et al. Catalyst design with atomic layer deposition [J]. ACS Catalysis, 2015, 5(3): 1804-1825.
[52] SARR M, BAHLAWANE N, ARL D, et al. Tailoring the properties of atomic layer deposited nickel and nickel carbide thin films via chain-length control of the alcohol reducing agents [J]. Journal of Physical Chemistry C, 2014, 118(40): 23385-23392.
[53] YAN H, CHENG H, YI H, et al. Single-atom Pd1/graphene catalyst achieved by atomic layer deposition: remarkable performance in selective hydrogenation of 1,3-butadiene [J]. Journal of the American Chemical Society, 2015, 137(33): 10484-10487.
[54] LU J L, STAIR P C. Low-temperature ABC-type atomic layer deposition: synthesis of highly uniform ultrafine supported metal nanoparticles [J]. Angewandte Chemie International Edition, 2010, 49(14): 2547-2551.
[55] LU J L, ELAM J W, STAIR P C. Synthesis and stabilization of supported metal catalysts by atomic layer deposition [J]. Accounts of Chemical Research, 2013, 46(8): 1806-1815.
[56] LU J L, FU B S, KUNG M C, et al. Coking-and sintering-resistant palladium catalysts achieved through atomic layer deposition [J]. Science, 2012, 335(6073): 1205-1208.
[57] LIANG X H, LI J H, YU M, et al. Stabilization of supported metal nanoparticles using an ultrathin porous shell [J]. ACS Catalysis, 2011, 1(10): 1162-1165.
[58] SUNG J, KOSUDA K M, ZHAO J, et al. Stability of silver nanoparticles fabricated by nanosphere lithography and atomic layer deposition to femtosecond laser excitation [J]. The Journal of Physical Chemistry C, 2008, 112(15): 5707-5714.
[59] LIU X Y, WANG A Q, ZHANG T, et al. Catalysis by gold: new insights into the support effect [J]. Nano Today, 2013, 8(4): 403-416.
[60] LIU X Y, LIU M H, LUO Y C, et al. Strong metal-support interactions between gold nanoparticles and ZnO nanorods in CO oxidation [J]. Journal of the American Chemical Society, 2012, 134(24): 10251-10258.
[61] YANG M, ALLARD L F, FLYTZANI-STEPHANOPOULOS M. Atomically dispersed Au-(OH)xspecies bound on titania catalyze the low-temperature water-gas shift reaction [J]. Journal of the American Chemical Society, 2013, 135: 3768-3771.
[62] FLYTZANI-STEPHANOPOULOS M. Gold atoms stabilized on various supports catalyze the water-gas shift reaction [J]. Accounts of Chemical Research, 2013, 47(3): 783-792.
[63] KWAK J H, HU J Z, MEI D, et al. Coordinatively unsaturated Al3+centers as binding sites for active catalyst phases of platinum on gamma-Al2O3[J]. Science, 2009, 325(5948): 1670-1673.
[64] YANG M, LI S, WANG Y, et al. Catalytically active Au-O(OH)x-species stabilized by alkali ions on zeolites and mesoporous oxides [J]. Science, 2014, 346(6216): 1498-1501.
[65] LI W Z, KOVARIK L, MEI D H, et al. A general mechanism for stabilizing the small sizes of precious metal nanoparticles on oxide supports [J]. Chemistry of Materials, 2014, 26(19): 5475-5481.
[66] GUO X G, FANG G Z, LI G, et al. Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen [J]. Science, 2014, 344(6184): 616-619.
[67] CAMPBELL C T, PEDEN C H F. Chemistry-oxygen vacancies andcatalysis on ceria surfaces [J]. Science, 2005, 309(5735): 713-714.
[68] FARMER J A, CAMPBELL C T. Ceria maintains smaller metal catalyst particles by strong metal-support bonding [J]. Science, 2010, 329(5994): 933-936.
[69] TA N, LIU J Y, CHENNA S, et al. Stabilized gold nanoparticles on ceria nanorods by strong interfacial anchoring [J]. Journal of the American Chemical Society, 2012, 134(51): 20585-20588.
[70] SI R, FLYTZANI-STEPHANOPOULOS M. Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2catalysts for the water-gas shift reaction [J]. Angewandte Chemie International Edition, 2008, 120(15): 2926-2929.
[71] LIN Q Q, HUANG Y Q, WANG Y, et al. RuO2/rutile-TiO2: a superior catalyst for N2O decomposition [J]. Journal of Materials Chemistry A, 2014, 2(15): 5178-5181.
[72] WANG A Q, LIU X Y, MOU C Y, et al. Understanding the synergistic effects of gold bimetallic catalysts [J]. Journal of Catalysis, 2013, 308(4): 258-271.
[73] PEI G X, LIU X Y, WANG A Q, et al. Ag alloyed Pd single-atom catalysts for efficient selective hydrogenation of acetylene to ethylene in excess ethylene [J]. ACS Catalysis, 2015, 5(6): 3717-3725.
[74] XU J, WHITE T, LI P, et al. Biphasic Pd-Au alloy catalyst for low-temperature CO oxidation [J]. Journal of the American Chemical Society, 2010, 132(30): 10398-10406.
[75] ZHANG H J, WATANABE T, OKUMURA M, et al. Catalytically highly active top gold atom on palladium nanocluster [J]. Nature Materials, 2011, 11(1): 49-52.
[76] ZHANG L L, WANG A Q, MILLER J T, et al. Efficient and durable Au alloyed Pd single-atom catalyst for the ullmann reaction of aryl chlorides in water [J]. ACS Catalysis, 2014, 4(5): 1546-1553.
[77] CHENG D J, HUANG S P, WANG W C. Thermal behavior of core-shell and three-shell layered clusters: melting of Cu1Au54and Cu12Au43[J]. Physical Review B, 2006, 74(6). DOI: 10. 1103/physRevB.74.064117.
[78] LIU X Y, WANG A Q, YANG X F, et al. Synthesis of thermally stable and highly active bimetallic Au-Ag nanoparticles on inert supports [J]. Chemistry of Materials, 2008, 21(2): 410-418.
[79] HE L, HUANG Y Q, WANG A Q, et al. Surface modification of Ni/Al2O3with Pt: highly efficient catalysts for H2generation via selective decomposition of hydrous hydrazine [J]. Journal of Catalysis, 2013, 298(1): 1-9.
[80] HE L, HUANG Y Q, LIU X Y, et al. Structural and catalytic properties of supported Ni-Ir alloy catalysts for H2generation via hydrous hydrazine decomposition [J]. Applied Catalysis B Environmental, 2014, 147(14): 779-788.
[81] CAO A, VESER G. Exceptional high-temperature stability through distillation-like self-stabilization in bimetallic nanoparticles [J]. Nature Materials, 2010, 9(1): 75-81.
[82] LI W J, WANG A Q, LIU X Y, et al. Silica-supported Au-Cu alloy nanoparticles as an efficient catalyst for selective oxidation of alcohols [J]. Applied Catalysis A: General, 2012, 433: 146-151.
Foundation item: supported by the National Natural Science Foundation of China(21203182, 21476226, 21506204).
Stabilization mechanism of supported metal catalyst
YANG Xiaoli, SU Xiong, YANG Xiaofeng, HUANG Yanqiang, WANG Aiqin, ZHANG Tao
(Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China)
Abstract:Supported metal catalysts play a pivotal role for many reactions in industrial processes, including petroleum refining, environmental protection and materials synthesis. However, the active metal centers suffer from severe sintering under real working condition, which will subsequently lead to a loss of its activity. Therefore, the stability of supported catalysts becomes a critical issue to its catalytic application. In this review, the aggregation and stabilization mechanisms of supported metal catalyst were briefly discussed according to the two different aggregation models of metal particles, i.e., Ostwald ripening and coalescence of smaller particles. Accordingly, the corresponding stabilization strategies, including physical methods based on encapsulation and chemical methods by forming chemical bonds, were introduced. The understanding of stabilization mechanism of supported metal catalyst would not only benefit for the development of new catalysts with promoted stability, but also provide theoretical guidance for the optimization of industrial catalysts.
Key words:supported metal catalysts; stability; aggregation; sintering; stabilization mechanism; stabilization strategy
Corresponding author:ZHANG Tao, taozhang@dicp.ac.cn
基金项目:国家自然科学基金项目(21203182,21476226,21506204)。
中图分类号:TQ 032.4
文献标志码:A
文章编号:0438—1157(2016)01—0073—10
DOI:10.11949/j.issn.0438-1157.20151556
