制备全系列聚烯烃材料的高性能单活性中心催化剂研究进展
2016-03-07艾则孜麦麦提明浙江大学化学工程与生物工程学院化学工程国家重点实验室浙江杭州310027
艾则孜·麦麦提明(浙江大学化学工程与生物工程学院,化学工程国家重点实验室,浙江 杭州 310027)
制备全系列聚烯烃材料的高性能单活性中心催化剂研究进展
艾则孜·麦麦提明
(浙江大学化学工程与生物工程学院,化学工程国家重点实验室,浙江 杭州 310027)
摘要:高性能单活性中心催化剂的发展给聚烯烃分子链结构的设计与“精确调控”提供了强大的工具,然而设计、制备和优化耐高温多功能催化剂成为制备高性能、高附加值聚烯烃产品的核心。本文回顾了全系列聚烯烃材料的种类,热性能和材料类型与共单体含量之间的关系。重点综述了:①改进型CGC-Ti、半茂钛、非茂铪(锆)等耐高温乙烯/α-烯烃共聚催化剂;②半茂钛、非茂钛等乙烯/(苯乙烯、降冰片烯)系列共聚催化剂;③优化型C2/C1-对称茂锆、耐高温非茂铪等高立构丙烯均聚和等规丙烯-乙烯系列共聚催化剂;④含杂环的C2/C1-对称茂锆高等规1-丁烯本体溶液聚合催化剂。与此同时,对这些催化剂的性能及其相应产物的链结构进行了详细地讨论。最后指出,优化耐高温等规型非茂铪和含杂环或杂原子的C2/C1-对称茂锆以及间规型C1-对称茂锆等多功能催化剂是今后的研究重点。
关键词:茂金属;非茂金属;催化剂;聚烯烃;制备;高通量催化剂筛选;链结构
第一作者及联系人:艾则孜·麦麦提明(1986—),男,硕士研究生,从事聚烯烃产品工程研究。E-mail 21128097@zju.edu.cn。
高性能单活性中心烯烃聚合催化剂的发展对聚烯烃链结构(分子量及分布、短支链及其链内和链间组成分布、长支链含量、立体构型及区域规整缺陷)的设计与精确调控提供了强大的机会;通过它们使定制无法由传统催化剂或机械共混等手段制造的新型高性能聚烯烃材料已经成为现实[1];负载型茂金属催化剂也拓宽了基于传统催化剂的聚合工艺可生产的产品范围[2]。ExxonMobil、Mitsui、Dow、LyondellBasell等公司用未负载单活性中心催化剂和高温溶液聚合工艺商业化开发了乙烯/(1-丁烯/1-辛烯)基、丙烯/(乙烯/1-丁烯)基和1-丁烯/(丙烯)基无规共聚物塑弹体以及乙烯基和丙烯基多嵌段共聚物热塑性弹性体(产品牌号分别为ExactTM/ VistamaxxTM、TafmerTM、EngageTM/InfuseTM/ IntuneTM、Koattro KT R05等)。然而传统单活性中心催化剂的高温催化性能不够理想:在特定聚合温度下制备高共单体含量产品时,催化剂效率和产品分子量明显下降[3](第二代聚烯烃弹性体——高分子量高熔体强度乙共聚物体需要用气相聚合工艺制造,产品牌号为EngageTMHM),丙烯基共聚物的立构规整度也明显降低[2]。因此,优化现有催化剂、设计并制备新型耐高温的高性能催化剂一直是开发工业化产品的难点之一[3-5],也是学术研究热点[6-9]。此外,筛选并优化高性能多用途催化剂,通过均相聚合方法调控烯烃基共聚物(由单体1和单体2及其相应共单体组成)的链结构而制备结构与性能均一的模型全系列材料——硬塑料1、软塑料1、塑性体1、弹性体1、橡胶1、软橡胶、橡胶2、弹性体2、塑性体2、软塑料2、硬塑料2(表1),系统地研究催化剂-聚合过程-链结构-加工过程-凝聚态结构与性能-应用之间的关系,从而实现聚烯烃树脂的高性能化、高附加值化也是极其迫切的。

表1 全系列聚烯烃材料的类型及部分结构与性能的关系
1 耐高温乙烯/α-烯烃共聚催化剂
通过单中心催化剂及聚合工艺调控聚烯烃产品的链结构,可以有效剪载其结晶结构与性能,从而拓宽其应用领域。①以Insite技术制备的乙烯/1-辛烯共聚物中辛烯摩尔含量与结晶形态、熔点、材料类型的关系:0%/大球晶-厚片晶/138℃/HDPE、2.8%/小球晶-薄片晶/118℃/LLDPE、5.2%/无带球晶-条纹晶和片晶的混合/84℃/塑性体、12.3%/缨状晶-粒晶/54℃/弹性体[25]。②长支链含量与加工性能和应用之间关系:长支链含量低的共聚物加工性能差,但适合于制备高强度薄膜、纤维、胶黏剂[3];长支链含量高的共聚物在高温下熔体黏度剪切变细、吹塑薄膜加工中气泡稳定性好,从而加工性能好,应用于电缆等领域[5]。③分子量及分布是选用何种加工工艺的最重要参数:第一代乙烯/1-辛烯共聚物弹性体作为聚丙烯相容剂而制备热塑性弹性体时,因其分子量较低,产品只能用注射成型加工,而第二代高分子量乙烯/1-丁烯共聚物弹性体熔体强度高,相应产品可以用热成型、挤出成型、吹塑成型工艺加工;分子量分布窄的聚烯烃弹性体在聚丙烯中分散性好,但作为材料使用时加工性能差[5]。
与非均相聚合工艺相比,单釜或多釜串联烯烃溶液聚合工艺更有利于定制链结构;与此同时,高温溶液聚合工艺有利于提高生产率、减少去挥发过程中能量消耗、降低结垢[3]。然而传统单中心催化剂的高温稳定性差,即当温度升高至特定温度以上(乙烯/α-烯烃共聚物:150℃[3];丙烯/乙烯共聚物:100℃[26])时,催化效率明显降低(产品含有催化剂残渣,不利于应用于医疗和电缆领域,需要进一步除杂,增加了生产成本)、共单体插入量变少、产品分子量降低(尤其是制备高共单体含量产品即聚烯烃弹性体时,分子量更低),见图1和表2中的C1和C7,只能下调乙烯转化率而提高产品分子量,从而降低了生产效率[3,5,27]。为此,有必要在优化现有催化剂的基础上设计并制备具有良好调控链结构能力的新型耐高温催化剂。
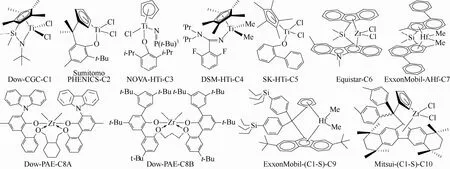
图1 工业应用高温乙烯/α-烯烃溶液共聚催化剂
1.1工业应用乙烯/α-烯烃共聚催化剂
第一代乙烯/α-烯烃溶液共聚催化剂C1~C6(图1)和rac-Me2Si(Ind)2HfMe2(Exxon,C7-H)的最大缺点是需要制备高共单体含量共聚物时,产品分子量低(表2,C1~C6)。因此,工业应用中只能降低乙烯和共单体转化率来提高产品分子量,然而这种方法导致生产率下降:Exxon公司用C7-H/([PhMe2NH][B(C6F5)4]/Al(n-OC)3[1∶(0.93~1.1)∶(32~34),摩尔比,下同]于高温高压(225℃,130MPa)连续溶液乙丁共聚所获得的产物中,丁烯质量分数为13.7%时,其MI(熔融指数)为3.02g/10min,而1-丁烯质量分数增高至25.4%时,产量无明显变化(分别为251kgPE/gCat、178kgPE/gCat),但MI降低到25.1g/min;高温中压(均为8.64MPa)连续溶液乙辛共聚中,其他条件相同而温度从142.2℃提高至160℃时,产物分子量相同(85kg/mol、84kg/mol,PDI为2.0~2.1),但辛烯质量分数从21.49%降低至16.30%,乙烯和辛烯转化率也分别从87.9%、54.9%调低到73.1%、33.1%[27]。
因此,Dow[3,26,29]、ExxonMobil[5,27]、Mitsui[4]等公司分别开发了制备立构型系列乙烯-丙烯共聚物的高性能多功能催化剂C8A/C8B、C7/C9、C10,其结构如图1所示。C8~C10在150~200℃的高乙烯转化率连续溶液聚合过程中,仍可以制备高共单体含量、高分子量乙烯基弹性体,如表2所示。
Dow化学公司用C8A/[HNMe(C18H37)2][B(C6F5)4]/MMAO-3A (1∶1.2∶5)在179℃、3.45MPa和以氢气作为分子量调节剂的条件下,乙烯转化率为91.2%时,仍获得MI为0.42g/10min、密度为0.868g/cm3的乙辛弹性体[29]。另外,C8B在脂肪烃中有良好的溶解能力:在20℃下在甲基环己烷中溶解度为10%,而C8A的只有1.6%;相同条件下C8B产生的长支链含量比C8A少[3]。
ExxonMobil化学公司用C9/[PhMe2NH] [B(C10F7)4]/Al(n-OC)3及高温(160℃)、高转化率(乙烯90.4%;丙烯56.3%)连续溶液共聚方法制备了符合应用于电缆的乙丙塑性体,产品中无残留催化剂及共催化剂,MIR(I21/I2) (表示长支链含量,聚烯烃塑弹体中其值在40~80之间)达到78.66;而用辛烯作共聚单体时,不能同时满足高活性、高I21/I2的要求,当乙烯和辛烯转化率为90.9%/59.9%时,I21/I2只为36.4[5]。
Mitsui石化公司的C10/MMAO/TIBA(1∶250∶2000)在聚合温度分别140℃和150℃并用氢气作为分子量调节剂的乙辛间歇溶液共聚过程中,得到高分子量(MI为0.79g/10min、2.8g/10min)、高辛烯含量(密度为0.865g/cm3、0.874g/cm3)乙辛弹性体,催化活性相当高(170kg/mmolZr、131.6kg/mmolZr);然而,温度升高至200℃而其他条件与表2的C10相同时,虽然产品辛烯含量和分子量基本不变(密度为0.905g/cm3,MI为7.3g/10min),但活性下降至35kg/mmolZr[4]。

表2 工业应用高温乙烯/α-烯烃溶液共聚催化剂的性能及其相应产物的部分链结构参数
1.2优化型限定几何构型催化剂
C1的最大优点是乙烯/α-烯烃共聚物的分子量不受共单体插入量的影响[35],而聚合温度在150℃以上时,催化效率下降、产品分子量降低(表2,C1);经研究发现其配体空间位阻和活性中心电子密度影响催化性能,研究者们优化其环戊二烯基、硅桥基、氨基、氨基取代基而制备了一系列CGC催化剂[36]。本部分只讨论优化环戊二烯基型、用苯基取代Si桥型、用邻苯氧基取代氨基型的高性能CGC催化剂,如图2所示的C11~C19。
RUCKENSTEIN等[35]比较了4种不同Cp、Cp*、Ind、2-Me-Benz(e)Ind (C11)基CGC的乙烯/辛烯共聚性能,其中C11/MAO的活性和辛烯插入量基本跟C1一致,而产物分子量高1.45倍;因此,他们详细研究了温度、共催化剂、溶剂对其性能的影响:溶剂对催化性能没有影响;随着Al/Ti比的增加,活性和分子量增大,而辛烯插入量保持不变;聚合温度从40℃升高至100℃时,活性提高了4倍,辛烯插入量不变,而从100℃升至160℃时,活性不变,辛烯插入量却增加了4.5%(摩尔分数,下同);其中80℃和160℃的性能列于表3。
VOSKOBOYNIKOV等[37]合成了2,3-二甲基茚基的5位或7位碳原子被硫原子杂化的两种CGC,前者表现出了更好的乙烯均聚和乙辛共聚能力(表3,C12)。GRANDINI等[38]合成了吲哚并茚和噻吩并茚衍生的多杂环CGC,其中C13/MAO在80℃下催化液相丙烯,得到高分子量、中等间规、无定形聚丙烯([rr]=52%,[rrrr]=71.2%,[2,1]<0.3%)(表3,C13);他们还研究了溶剂丙烯聚合中共催化剂、丙烯压力、温度对催化性能的影响:共催化剂的活性顺序为[Ph3C][B(C6F5)4] > MAO =[PhNHMe2] [(C6F5)4],对产物间规度影响不大,但MAO产物的分子量较低;C13/[Ph3C][B(C6F5)4]的活性跟丙烯浓度呈一级依赖关系,而间规度和分子量没有明显变化;随温度升高,催化活性、间规度、产物分子量都随之下降。

图2 乙烯基共聚催化剂
MILLER 等[39]合成了一种新型CGC-Zr多功能催化剂C14,在0℃下得到高间规聚丙烯(见3.2节);C14/MAO在75℃和0.54MPa下,对乙烯和α-烯烃表现出了跟C1/MAO完全不同的催化性能:在相同条件下,按单体浓度校正的乙烯均聚活性比1-辛烯均聚活性低6倍(C1,90倍),比4-甲基-1-戊烯均聚活性高2倍(C1,140倍);乙烯/α-烯烃共聚活性与C1相当,并与共单体插入率一样随共单体用量的增加而呈线性比例增加(包括纯共单体),1-辛烯/4-甲基1-戊烯共聚活性比C1高52倍。他们还用乙烯为唯一单体成了含乙烷短支链和长于正己烷的长支链(但不含C1/C3/C4/C5)的LLDPE,但其分子量很低。因此,为了研究茐基6、7位取代基对聚乙烯分子量、短支链和长支链含量的影响,他们制备了带1,1,4,4-四甲基-7-R-1,2,3,4-四氢化苯并茐(R:H、Et、tBu,分别为C14-H、C14-E、C14-B)和C15的Si(Me)2桥架被Si(Ph)2取代的C14-Ph,结果C14-E不产生支链,而其他3种催化剂产生的短支链含量基本相同(3.3~4),长支链含量顺序为C14-H(17.2)>C14-B(14.9)>C14-Ph(9.1),产物熔点分别为127℃、123℃、123℃;分子量(分布)为382.1(86)kg/mol、472.1(30)kg/mol、33.6(17)kg/mol、7.4(2.5)kg/mol;40℃下,C14-Ph的乙烯/1-十二辛烯共聚过程中,共单体相对活性相当高,rD=0.49,rE=14.9,rE·rD=0.73[40]。
LEE等[41]合成了由2-R-1,2,3,4-四氢喹啉衍生物(R=甲基、正丁基、叔丁基、叔丁基醚己基)构造的邻苯桥连(四甲基环戊二烯基、茚基、茐基)/氨基钛催化剂。其中C15/([Ph3C][B(C6F5)4])/TIBA的乙烯/辛烯共聚活性比C1高13.6倍(表3),辛烯插入量多1.32倍、分子量低1.4倍。他们又合成了o-(p-R1-Ph) (Cp*/Ind/Flu)/(R2-N)-Ti-Me2[R1=H、Me; R2=Me、Et、CH2(CH2)2CH3、CH2C(Me)3、CH(Me)2]催化剂,其中o-Ph-Cp*-N(Et)-Ti-Me2(C16)乙辛共聚性能接近于C1;相同条件下,活性低2.6倍,而产物分子量高2.3倍,辛烯插入量持平[42]。TAO等[43]合成了o-PhC-Cp*-N(R)-Ti-Cl2(R=iPr、Cy、nPr、4-MePh),其中氨基取代基为Cy的C17具有较强的乙烯均聚和乙烯/1-己烯共聚能力(表3,C17),其产物中己烯在分子链内分布趋于交替(rE·rH=0.2)。

表3 耐高温乙烯/(1-辛烯、1-己烯)共聚催化剂的性能
Sumitomo 化学公司的Imai A等合成了系列[{Me2C(/Si)-(Cp/Cp*)(OC6H2-6-R1-4-R2)}TiCl2](碳桥:R1=H、tBu,R2=H、Me;硅桥:R1=iPr、tBu、Adm,R2=Me)(简称为PHENICS),其中C18在 80℃下的乙烯/1-己烯共聚活性比C1高26倍,其他性能基本一致;而高温高压(180℃,2.5MPa)下,活性、产物分子量及己烯含量与低温低压(80℃,0.6MPa)相比,分别下降了4.5倍、3.2倍、1.31倍, 如表3所示的C18;在80℃和0.6MPa下,[{Me2C-(Cp)PhO}TiCl2]/[Ph3C][B(C6F5)4]/TIBA的A1/Ti比对己烯插入量影响很大,A1/Ti为50、125、500时,活性基本不变[分别为2.8kg/(mmol·h)、1.8kg/(mmol·h)、2.07kg/(mmol·h)],但己烯插入量大幅度降低(摩尔分数分别为34.6%、28.1%、8.9%)[30]。他们再通过优化环戊二烯基(如Cp/Cp*/Ind)和苯氧基邻位取代基(tBu,iPr),实现了对乙烯/α-烯烃共聚物分子量的有效调控,并发现产生高分子量产物的根本原因是此类催化剂能够有效抑制α-烯烃的[2,1]插入方式;因此该公司的HANAOKA等[6]合成了单噻吩并和双噻吩并环戊二烯基系列PHENICS催化剂,并用Symyx公司的高通量筛选方法在低压(0.6MPa)和不同温度(40℃、70℃、130℃)下进行筛选,发现他们乙己共聚行为均超过[{Me2Si-(Cp*)(OC6H2-6-tBu-4-Me)} TiCl2],其中C19(图1)在超高温、高压下仍保持优良的乙烯/1-己烯共聚能力(表3,C19)。
1.3半茂钛系催化剂
NOVA[31,46]、DSM[32]和NOMURA K[44-45,48-49]等分别开发了以C3/C22、C4、C20-23/C32-34为代表的半茂钛催化剂,其结构如图1和图2所示。在MAO的引发下,通过优化环戊二烯基和阴离子配体而控制其金属活性中心的电子密度和空间位阻,不仅高效催化乙烯/(α-烯烃、乙烯基环己烷、3-甲基-1-戊烯)共聚,还可以制备高分子量、高共单体含量的乙烯/(苯乙烯、2-甲基-1-戊烯、环烯烃)共聚物;其乙烯/α-烯烃共聚性能列在表3中。
NOMURA等[44-45]考察了环戊二烯和苯氧配体中的取代基对乙烯均聚和乙烯/α-烯烃共聚性能的影响,其中C20~C21在低温下可以制备无规立构型乙烯-(α-烯烃)全系列共聚物。值得一提的是,当共单体用量增加时,催化剂活性和产物分子量均增加,并且共聚单体在链内组成分布趋于交替结构(C20:rE·rH=0.29,nE=2.7,nB=0.11;C21:rE·rH=0.41,rE=2.35,rH=0.18)。在高温高压连续溶液过程中,NOVA公司的C22A比C22B表现出更好的乙辛共聚能力,但乙烯转化率只有60.3%[46]。KIM等[47]合成了苯氧基的2,6-位双取代和邻位单取代的系列半茂钛催化剂,并用高温半连续溶液聚合方法评价了乙烯均聚和乙烯/1-辛烯共聚性能,发现C5的活性、产物分子量和辛烯含量都高于其他同类催化剂及C1(表3,C5);高温连续溶液动力学实验也表明C5的辛烯插入速率和含量都高于C1。NOMURA 等[24]合成的C23在低温下能制备高己烯含量(47.6%摩尔分数)、超高分子量(Mw=1947kg/mol)乙己共聚物,其己烯的链内组成分布趋于无规(rE·rH=0.63,rE=9.56,rH=0.066),说明乙烯/己烯的相对活性差距大于C20,因而其乙烯/1-十二辛烯共聚物中1-十二辛烯摩尔分数只为3.6%。
1.4基于高通量催化剂筛选技术开发的非茂铪催化剂
相比于传统催化剂研究方法,高通量催化剂筛选技术不仅具有迅速识别活性催化系统的能力,更重要的是能识别并抛弃无催化活性系统,而且可以评估更广泛的催化剂候选者和反应条件。因此,该技术更适合于易合成的非茂配体及其催化剂的发现和优化。因此,Dow化学公司和Symyx技术公司合作发现了高性能胺醚基和酰胺醚基铪乙烯/α-烯烃共聚催化剂(C24~C25,图2)[7]、耐高温吡啶酰胺基[9]和咪唑胺基铪[9]高等规丙烯聚合催化剂(C42/C46,图3)及多功能桥连芳醚铪(锆)催化剂(C45,图3)[26]。
乙烯/α-烯烃共聚催化剂筛选步骤一般分为初选、二次筛选和三次筛选。初选主要是发现潜在的新配体和金属的组合,作为具有特定应用价值的聚合催化剂,用辛烯均聚进行评价;二次筛选为迅速优化初选催化剂,较大规模和工业应用条件下进行乙烯/1-辛烯共聚,建立催化剂配体结构与活性、分子量及辛烯插入率之间的关系;三次筛选即传统间歇釜方法评价并确认二次筛选过程中优化的催化剂。通过此过程筛选的C24、C25乙烯/辛烯共聚性能列于表4:相同条件下,C25的共聚活性比C1低3.8倍,但产物分子量比C1高3.04倍[7]。
Union Carbide公司开发的第一代亚胺酰胺类催化剂引起了Dow化学公司研究人员的兴趣。他们与Symyx技术公司合作通过高通量筛选技术发现了第二代亚胺-酰胺类催化剂——吡啶-酰亚胺铪[9]。然而,亚胺-酰胺类催化剂高温催化乙烯/α-烯烃共聚时,发生异构化或分解,从而表现出多活性中心特点,如图3和表4所示的C44[50-53]。因此,为了提高该类催化剂的高温稳定性,他们设计并制备了C26~C28(图2),其乙辛共聚性能列于表4[51-53](其中C26-Bn3的收率很低,因而他们改进合成方法制备了可放大的C26[51]);C27高温下表现出多种活性中心特点,其产物经GPC和DSC表征:分子量呈双峰分布,有两个80.2℃和106℃熔点[52]。
Mitsubishi化学公司与Coates合作开发了C29(图2),并在90℃下乙烯/α-烯烃与立构大单体共聚制备了支化嵌段共聚物,其中分子量为26.9kg/mol的等规聚丙烯大单体插入量为8.8%(质量分数),主链中α-烯烃摩尔分数为34%[54]。
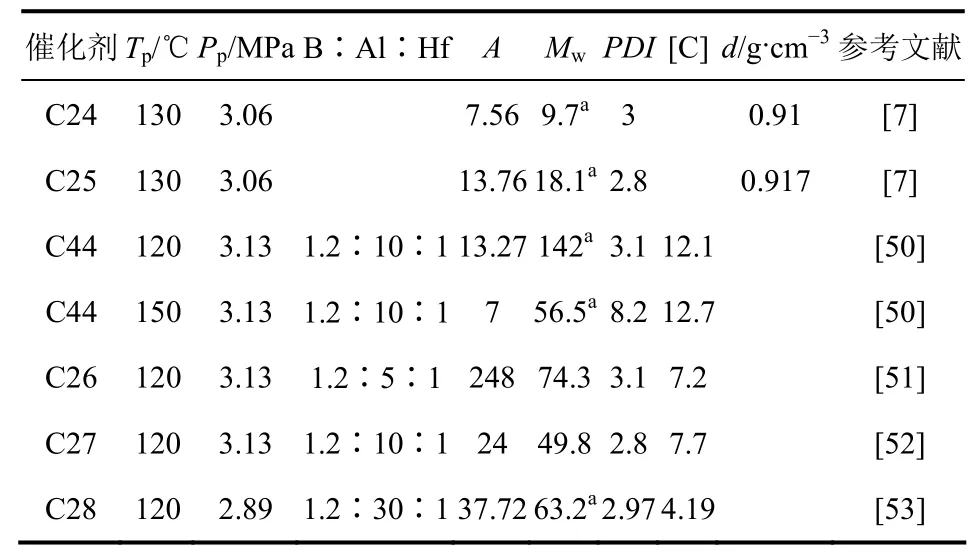
表4 亚胺-烯胺系列非茂铪系催化剂的高温乙烯/1-辛烯共聚性能
2 制备新型乙烯基共聚物的多功能催化剂
2.1间规立构型苯乙烯-乙烯系列共聚催化剂
间规聚苯乙烯是一种具有高熔点(270℃)的工程塑料,只能通过CpTiCl3等半茂钛催化剂配位聚合得到,而通过该类催化剂乙烯/苯乙烯共聚时,得到聚乙烯、乙苯共聚物和间规聚苯乙烯的混合物[49]。用CGC催化乙烯/苯乙烯共聚时,扰乱聚乙烯结晶结构而制备从塑料到玻性体的一系列材料(表1)。因此,需要设计并制备具有同时高效催化苯乙烯间规均聚和乙烯/苯乙烯无规共聚的多功能催化剂。
KLOSIN等[52-53]对C1进行乙烯/苯乙烯共聚动力学研究时发现,若在增长的分子链中倒数第二位存在苯乙烯(不是乙烯),会显著增加反应速率,即“倒数第二苯乙烯效应”。他们就假设如果活性中心附近的取代基被芳基取代时,可能会增加苯乙烯插入速率。因此他们合成了环戊二烯基3,3'-苯基取代的两种催化剂,结果苯乙烯的插入率及产物分子量明显提高,可以满足商业化溶液聚合工艺,如表5所示的C30。
NOMURA等[49]合成双核CGC(C30,图2),制备了分子量为8kg/mol的无规聚苯乙烯,而乙烯/苯乙烯共聚时得到高分子量、高苯乙烯含量共聚物(表5,C31A),然而活性较低。NOH等[56]合成了分别以苯、对二甲苯、对二乙基苯链接的双核CGC,其中以对二乙苯链接的C31B比C1和其他同类催化剂具有更好的乙苯共聚性能,然而分子量明显低于C31A(表5,C31B)。
NOMURA等[24]发现了同时具有高效催化乙烯/苯乙烯无规共聚(C21~C23,图2)和苯乙烯-乙烯系列共聚(C20、C32~C33,图2)的半茂钛催化剂,其性能列在表5里(序号4~10为乙苯无规共聚,序号11~13为苯乙烯间规均聚)。其中C22A对乙烯/苯乙烯共聚表现出活性共聚特征,但没有活性催化它们的均聚反应;C23跟乙己共聚一样得到超高分子量乙苯共聚物;虽然C20高温下能得到高分子量间规聚苯乙烯,但是较高温度下得不到均相乙烯/苯乙烯共聚物。为此,他们通过优化催化剂、共催化配比,获得了均相高苯乙烯含量乙苯共聚物(表5,C20A),对实际应用具有重要意义[47]。

表5 双核CGC及半茂钛催化剂的乙苯无规共聚(1~10)及间规苯乙烯均聚(11~13)性能
一般认为,活性中心Ti的价态Ti(Ⅲ)和Ti(Ⅳ)分别负责催化苯乙烯间规均聚和乙烯/苯乙烯无规共聚。为了阐明半茂钛催化剂的乙烯-苯乙烯系列共聚机理,NOMURA等[49]用C33/C21二步聚合,即第一种是先苯乙烯聚合再加入乙烯,第二种是先乙苯共聚再抽掉乙烯。前过程中,两种催化剂都可以得到间规聚苯乙烯和乙苯共聚物的共混物,并且催化性能跟单独聚合过程没有区别,而后过程中,则没有间规聚苯乙烯生成,结论是苯乙烯间规均聚活性中心Ti(Ⅲ)可以转化为乙苯共聚活性中心Ti(Ⅳ),反之则不能。
乙苯共聚物中苯乙烯插入方式随催化剂结构的不同而不同,C1催化的苯乙烯在链中以区域和立体不规则插入方式增长,产物中存在以尾-尾链接结构;双核CGC产物苯乙烯摩尔含量大于50%时存在头-尾三元结构和尾-尾二元结构;半茂钛催化剂产物中除了存在头-尾链接的二元、三元结构外,还存在由两个乙烯隔开的头-头和头-尾链接结构[48]。
2.2乙烯/降冰片烯共聚催化剂
乙烯/降冰片烯共聚物具有高透明、耐高温、高折射率等独特特性,在光学材料方面具有潜在的应用价值,如光盘、透镜、光纤、薄膜等。通过C2-[19-20]/C1-[20]/Cs-对称[20]茂锆催化剂、CGC[57]和一些非茂钛催化剂[58]已制备了等规/无规聚降冰片烯、交替/无规共聚物、嵌段共聚物,并通过控制乙烯或丙烯的插入量调控其玻璃化温度而改善了加工性能。
然而,多数催化剂的性能随降冰片烯用量的增加而下降。与此相反,半茂钛、非茂前过渡催化剂显示出了较好的共聚性能,如表6所示的C22B和C34~C37,其结构如图2所示。其中半茂钛催化剂C22B和C34的活性和产物分子量较高,降冰片烯链内组成分布分别呈无规和交替,而且存在三元重复结构单元[45,59]。相同条件下,C36的共聚活性跟C1基本一致,但降冰片烯插入量高1.36倍,其立体构型为无规(间规/等规=55/45),链内组成分布为交替(交替率为96.1%),含量不随用量的增加而增多;这种催化剂最大的特征是在5min内得到高分子量(198kg/mol)和窄分布(PDI=1.13)的共聚物(Tg=129℃),其活性聚合特征有利于制备高性能嵌段共聚物[57]。
2.3制备新型乙烯/(大位阻α-烯烃)共聚物的催化剂
NOMURA等[59]发展的半茂钛催化剂提供了制备无法通过其他单中心催化剂而获得新型乙烯/(环己烯、2-甲基-1-戊烯、叔丁基乙烯、含硅共单体)共聚物的新途径。它们的环戊二烯基和阴离子配体结构对共单体的选择性有较大的影响,其中若干个高效催化剂(如图2所示的C20-23/C31A)的性能列在表6。此外,他们用C20和C22B得到了乙
烯基环己烷插入量分别为31.9%(摩尔分数,下同)和24.5%的高分子量(230kg/mol、812kg/mol)乙烯/VCH共聚物,C20的活性[14.1kg/(mmol·h)]比C22B高两倍[62]。用C21和C33也可以得到环己烯插入量分别为16.1%和14.1%的乙烯/CHE共聚物,其中C33具有中等活性[1.66kg/(mmol·h)],产物分子量也较高(83.6kg/mol)[63]。

表6 乙烯/降冰片烯共聚及新型乙烯/(大位阻α-烯烃)共聚催化剂的部分性能
2.4制备交替乙烯/α-烯烃共聚物的催化剂
作为轮胎材料的天然橡胶链结构中,存在完全交替的乙烯/丙烯组成单元,但具有双键而不耐热和光,因此用乙烯和丙烯两种大宗单体制备交替乙丙橡胶(饱和异戊橡胶),可以提高异戊橡胶的耐老化性,而且能降低生产成本;此外,如果能控制丙烯的立体构型,可以创造新型高性能乙丙橡胶。
具有交替对映配位增长中心的C1-对称催化剂的发展使高效制备乙丙交替共聚物成为可能。研究人员通过[{(3-MeCp)(Me2C)(Flu)}ZrMe2]、[Me2Si (Ind) (Flu)]ZrCl2和[Me2Si(4,7-Me2Ind) (Flu)]ZrCl2分别得到了半等规、中等等规、间规聚丙烯和中等三元等规交替结构、低分子量无规交替、高分子量无规交替乙丙共聚物[64]。因此,KAMINSKY等[64]比较了4个系列C1-对称茂金属催化剂[X2Si{(2-Y)Ind)} (Z)]ZrCl2(X=Me、Ph;Y=H、Me;Z=茐基、2,7-二叔丁基茐)、C38和Me2Si-C38的交替乙丙共聚性能(温度为0℃、15℃、30℃、45℃、60℃)。其中,[Me2Si(Ind) (2,7-tBu2Flu)]ZrCl2在0℃下,乙丙共聚物乙丙交替率最高(EPE+PEP=85.5%),但在60℃下迅速降至61.2%,而且分子量很低(14.9~19kg/mol)、玻璃化温度为−65.3~−60.1℃,催化剂活性在0.48~40.46kg/(mmol·h);与此相反,C38产品在15~45℃乙丙交替率不变(75%~74%),在60℃下则下降至68.1%,催化活性为0.193~82.18 kg/(mmol·h),其产物玻璃化温度为−54.8~−53.8℃、分子量为150~160kg/mol、三元交替单元的立体构型趋于等规,而其聚丙烯均聚物立体构型则为中等间规。在30℃下单体浓度对交替率有较大影响:从1mol/L升高至4mol/L时,Me2Si-38产物乙丙交替率从65%增大到75%,其合理解释是反跳聚合机理(back-skip)导致在低单体浓度下增加非交替序列。UOZUMI等[65]用Et(1-Ind)(9-Flu)ZrCl2-MAO在0℃下制备了交替率接近100%([EC]>99%;[ECE]+[CEC]>98%)的乙烯/1-葵烯共聚物,分子量为35.2kg/mol,玻璃化温度为−68.4℃。KAMINSKY 等[66]用C39/MAO在30℃下也制备了高分子量(Mv=530,黏均相对分子质量)乙辛共聚物,但交替率为70%([EEE]=15.07%,[EOE]=63.8%,[OEO]=31.36%)。
3 高立构丙烯聚合催化剂
相比于乙烯基共聚物,基于单中心催化剂的聚丙烯产品没有大规模工业化[2],其重要原因是缺乏具有同时调控分子量、立体构型([mmmm])与区域规整缺陷([2,1]、[3,1])能力的易合成催化剂。虽然第一代等规丙烯聚合催化剂ansa-[Me2Si(2-Me-4-Ph-Ind)2]ZrCl2(AZr-1)[67]和CS-对称丙烯间规聚合催化剂[Me2C(Cp)(Flu)]ZrCl2一般条件下可以制备高分子量、高立构聚丙烯,但在70℃以上聚合温度时,其产物分子量和立构规整度大幅度下降,更不适合应用于新型高效超临界液相丙烯聚合工艺和高温溶液聚合工艺,因此需要优化现有催化剂,设计并合成新型耐高温高立构丙烯聚合催化剂。
3.1高等规丙烯聚合催化剂
3.1.1优化型C2-对称茂锆催化剂
EWEN等[67]合成了一系列了噻吩并和吡咯并环戊二烯基手性柄型茂锆催化剂。在无氢气作为链转移的液相丙烯聚合中,催化剂配体的坠儿苯中没有取代基时活性最高(比AZr-1高3.77倍),而有双取代基时(C40,图3)分子量最高(比AZr-1 低1.49倍),产物熔点为160℃,链结构缺陷均低于其他催化剂([mrrm]=0.26%,[2,1]=0.24%);氢气存在下,C40产物中无规聚丙烯(aPP)含量为0.1%(无氢:0.9%), 其不同助催化剂活化时的性能列于表7;在AliBu3/[Ph3C][B(C6F5)4]/H2催化下,坠儿苯邻位只有一个甲基取代时,得到超高分子量聚丙烯(1165kg/mol)。
SCHOBEL[68]合成了C41(图3),并在TIBA/ [Ph3C][B(C6F5)4]和溶剂丙烯聚合条件下,考察了温度和压力对催化性能的影响:在0℃下得到超高等规、超高分子量聚丙烯,其熔点为171℃;在0~30℃下链中不存在可检测的结构缺陷;在50~90℃下,[2,1]含量为0.06%~0.01%,[3,1]含量为0.02%~0.15%,活性增高而产物分子量降低(表7);在70℃下,压力从0.4MPa提高至0.6MPa时,[3,1]含量减少2倍,而[2,1]含量不变,活性和产物分子量分别增大1.46倍和1.64倍。

图3 高立构丙烯聚合及等规丙-乙共聚催化剂
3.1.2耐高温C2-对称茂锆催化剂
NIFANT’EV等[8]合成了AZr-1茚基的5,6-位氢分别被OMe和叔丁基取代的C56(图3),在120℃下得到高分子量、高等规聚丙烯 ([mmmm]= 93.8%,熔点为147.8℃,分子量为298kg/mol),与100℃条件(表7,C56)相比,活性只下降了7.92倍。其原因是OMe对阳离子活性中间体具有稳定化作用,因而下降的Zr有效电荷可能抑制通过元结转化态(agostic transition state)发生的β-消除以及β-H向单体链转移过程。
3.1.3C1-对称茂锆催化剂
MILLER等[69]合成的C47中,2-甲基2-金刚烷基和八甲基八氢化二苯并茐基分别促进端异构化(site epimerization)和分子链直接从单体插入过渡态增长的同时,还共同提供特定的单体插入端以提高立构规整度。因此,在0℃下得到熔点为167℃的高等规度聚丙烯。在20℃下,虽然熔点下降至163℃,但是催化剂活性和产物分子量分别提高了2.41倍和1.15倍(表7,C47)。
KIRILLOV等[70]合成了C1-对称催化剂C48~C49(图3),相比于C2-对称茂锆,该类催化剂产物的分子量较低(表7,C48~C49)。当聚合温度为60℃时,两个催化剂产物中β-H或β-Me消除而形成的乙烯基/亚乙烯基/异丁烯基含量都小于2%,而在100℃时,其含量分别42/46/12和32/68/0。3.1.4基于高通量筛选技术开发的非茂铪催化剂
Dow化学公司与Symyx技术公司合作基于高通量催化剂筛选技术开发的C42(图3)[9],在氢气存在下能制备高分子量聚丙烯,但等规度不太高[71](立体构型缺陷:[mr]=5.79%,[rr]=2.08%),经优化的C43产物具有较高的等规度和熔点(立体构型缺陷:[mr]=4.45%,[rr]=1.62%)(表7,C42~C43)。
Dow化学公司等研究了C44(图3)和其对应Zr化合物的丙烯聚合机理:Zr化合物的活性和分子量比C44-Hf低1倍和3.12倍,产物等规度稍低一点。聚合温度为120℃时,C44产生较高的[2,1]错位插入量,产物分子量和熔点都明显下降(表7,C44)[50]。
LAPOINTE等[9]通过高通量筛选发现并优化了噻唑/咪唑-酰胺基铪催化剂,其中C46(图3)在130℃下活性比C44高16.25倍,而其他性能较接近,其活性和产物的四元等规结构含量([mmmm])分别约为5000mg/(umol·min) 、95%。

表7 高等规丙烯聚合催化剂的性能
3.2高间规丙烯聚合催化剂
MILLER等[72]合成了C50和C14A(C14无乙醚配位的化合物)(图3)。其中C50在0℃下5min内得到高分子量(495.4kg/mol)、高间规聚丙烯,但不耐高温,即在90℃下只得到中等间规、无定形聚丙烯; 与此相反,此条件下,C14A的产物间规度和熔点都高于C50,但分子量较低(表8,C50/C14A)。
FINA公司开发的C51(图3)在−10℃下得到了[rrrr]值为90.7%而熔点到目前为止最高的间规聚丙烯,如表8所示;更重要的是,该催化剂工业应用条件下仍能得到高间规、高分子量产物[73]。

表8 间规丙烯均聚(C50~C51,C14A)及等规丙烯/乙烯共聚(C53~C56)催化剂的性能
3.3制备高分子量等规丙-乙系列共聚物的催化剂
虽然通过C7、C52(图3)得到高分子量、高等规聚丙烯,但丙烯/乙烯共聚制备丙乙无规共聚物或塑性体时,产品分子量和活性大大降低,无法满足工业应用要求[70,75]。
Basell聚烯烃公司[74]和BAGROV等[76]分别开发的C53/C55、C54(图3)对丙烯均聚和丙烯/乙烯共聚具有高效催化特性(如表8所示)。其中未负载的C54在液相丙烯本体聚合中,以氢气作为链转移时,活性提高了4.91倍,产物熔点为167.9℃;用硅胶/MAO负载时,活性和等规度明显下降(表8,C54),氢气用量对催化性能有较大的影响:随着其用量的增加,活性先增加到最高值再下降。用二步聚合方法制备聚丙烯合金时,橡胶相乙烯质量分数为8.5%~57%,活性为14.3~65.6kg/(mmol·h),产物特性黏度基本不变。
NIFANTEV等[8]设计的C56(图3)不仅在120℃下合成高等规、高分子量聚丙烯,而且在100℃下全乙烯浓度范围丙烯/乙烯共聚时,催化剂性能基本保持一致(表8,C56)。
Dow化学公司的C43/PMAO-IP/B(C6F5)4(1∶30∶1.2,见图3)在连续本体丙烯/乙烯共聚过程(80℃,3.4MPa)中,丙烯/乙烯的转化率为48%/98.7%时,在氢气存在下得到高分子量(Mw=306.8kg/mol,PDI=2.4)、高等规([mm]=95.1%)丙乙共聚物,乙烯含量为18.6%(摩尔分数),区域规整缺陷含量为0.37%([mr]=0,[rr]=4.5%),催化效率为278.6kg/gCat[71]。
4 高等规1-丁烯聚合催化剂
高等规聚1-丁烯是一种产量低而附加值很高的高性能绿色工程塑料,应用于高性能管材领域。因为该树脂在40℃以上温度时溶于液相1-丁烯单体,从而其液相本体溶液聚合工艺可以直接用未负载的单中心催化剂而无需考虑其颗粒形态。一般而言,高立构聚丙烯催化剂能催化1-丁烯生成相对更高立构的聚1-丁烯,但分子量和活性都下降;因此很长一段时间内没有一种具有工业化应用价值的催化剂被开发出来[77]。
Basell公司的Resconi等[77]研究了C2/C1-对称茂锆催化剂对液相本体1-丁烯聚合性能。其中C2-对称茂锆M-Zr ([C(3-tBuInd)2ZrCl2])产物的[mmmm]很高,但分子量很低。催化剂C40和C57(图3)在60~90℃范围内表现了优良的综合催化性能:聚合物分子量和熔点没有随温度的升高而明显下降。这些催化剂的链结构缺陷差别很大:催化剂M-Zr的产物[mmmm]跟ZN基本相同,但链结构缺陷([mr]、[rr])含量比ZN大,因而熔点低于ZN产物;C44产物的[mmmm]为100%,但存在较高的异构插入含量,导致熔点明显低于ZN;C57产物的[mmmm]较低,并且[2,1]含量较高,但是熔点接近于前两者,如表9所示。因此,针对C57的良好催化性能,他们研究了氢气用量对其活性和聚合物分子量的影响,发现C57对氢气具有良好的敏感性,在低Al/Zr=200和氢气存在下,仍保持高活性[220 kg/(g·h)]的同时,产物Mv高达245.6。
ROSA等[13]用C57合成了乙烯摩尔含量在0~28.62%的丁乙共聚物:随着乙烯含量的增加,黏均相对分子质量从229.3增高至532.6,分子量分布在2.1~3.1之间,玻璃化温度从−31.3℃降低至−50.6℃,物理性覆盖硬塑料、软塑料、塑性体和弹性体。

表9 高等规丁烯-1本体溶液聚合催化剂的性能及产物链结构缺陷[77]
5 结语与展望
(1)苯氧基优化氨基的CGC和基于高通量筛选技术的亚胺-烯胺基铪系催化剂具有高温乙烯/高级α-烯烃溶液共聚能力;基于高通量筛选技术吡啶酰胺和咪唑胺基铪系催化剂能得到高等规聚丙烯及高分子量丙乙共聚物,然而在高温下其乙烯/α-烯烃共聚物链结构不均匀,因此需要充分利用分子模拟和高通量筛选技术,优化此类催化剂,设计、发现并制备具有定制乙烯-丙烯全系列共聚物的新型多功能催化剂。
(2)部分半茂钛催化剂(包括含苯-X基(X=O、S、P )的非茂钛)在中、低温下可以制备乙烯-(α-烯烃、环烯烃、苯乙烯)全系列共聚物,但需要优化其配体结构、共催化剂的类型及配比,使之高温下得到均相系列材料。
(3)C1-对称茂锆催化剂和基于高通量筛选技术的多芳醚铪(锆)系催化剂不仅具有高效催化超高温乙烯/(α-烯烃、苯乙烯)溶液共聚的能力,而且能很好地控制产物长支链结构,从而应用于不同领域,而且还可以制备高立构丙烯-乙烯全系列共聚物,但需要优化其结构,在高温下控制聚丙烯立体构型和区域规整缺陷,从而制备高熔点丙烯基材料。
(4)改进型C2-对称茂锆催化剂可以在高温下制备高等规、高分子量丙-乙全系列共聚物,然而高分子量、高等规丁-丙全系列共聚物只由C2-对称噻吩茂锆催化制备。因此,需要进一步优化并制备含氧和硫原子的杂化催化剂,高温下制备高等规、高分子量丙烯-乙烯和丁烯-丙烯全系列共聚物。
参考文献
[1] CHUM P S,SWOGGER K W. Olefin polymer technologies——history and recent progress at the Dow Chemical Company[J]. Prog. Polym. Sci.,2008,33:797-819.
[2] SINCLAIR K B. Future trends in polyolefin materials[J]. Macromol. Symp.,2001,173:237-261.
[3] Dow Global Technologies INC. High temperature polyethylene solution polymerization process:US2012/0108770A1[P]. 2012-05-03.
[4] Mitsui Chemicals INC. Process for producing olefin polymers:US2006/0270812A1[P]. 2006-11-30.
[5] ExxonMobil Chemical Company. Process for making ethylene interpolymers and interpolymers made thereby:compositions and electrical devices containing such interpolymers:US2005/0215737A1 [P]. 2005-09-29.
[6] SENDA T,HANAOKA H,OKADO Y,et al. Titanium complexes of silicon-bridged cyclopentadienyl-phenoxy ligands modified with fused-thiophene:synthesis,characterization,and their catalytic performance in copolymerization of ethylene and1-hexene[J]. Organometallics,2009,28:6915-6926.
[7] BOUSSIE T R,DIAMOND G M,GOH C,et al. A fully integrated high-throughput screening methodology for the discovery of new polyolefin catalysts:discovery of a new class of high temperature single-site group (Ⅳ) copolymerization catalysts[J]. J. Am. Chem. Soc.,2003,125:4306-4317.
[8] NIFANT’EV I E,IVCHENKO P V,BAGROV V V,et al. 5-Methoxy-substituted zirconium bis-indenyl ansa-complexes:synthesis,structure,and catalytic activity in the polymerization and copolymerization of alkenes[J]. Organometallics,2012,31:4962-4970.
[9] DIAMOND G M,HALL K A,LAPOINTE A M,et al. High-throughput discovery and optimization of hafnium heteroaryl-amido catalysts for the isospecific polymerization of propylene[J]. CS Catal.,2011,1:887-900.
[10] CHEN H Y,CHUM S P,HILTNER A,et al. Comparison of semicrystalline ethylene–styrene and ethylene–octene copolymers based on comonomer content[J]. Polym. Sci.,Part B:Polym. Phys.,2001,39:1578-1593.
[11] XU G X,CHENG D L. Homo- and copolymerization of 4-methyl-1-pentene and ethylene with group 4 ansa-cyclopentadienylamido complexes[J]. Macromolecules,2001,34:2040-2047.
[12] STEPHENS C H,POON B C,ANSEMS P,et al. Comparison of propylene/ethylene copolymers prepared with different catalysts[J]. J. Appl. Polym. Sci.,2006,100:1651-1658.
[13] ROSA C D,ODDA B R D,AURIEMMA F,et al. Polymorphic behavior and mechanical properties of isotactic1-butene-ethylene copolymers from metallocene catalysts[J]. Macromolecules,2014,47:4317-4329.
[14] ROSA C D,AURIEMMA F,VOLLARO P,et al. Crystallization behavior of propylene-butene copolymers:the trigonal form of isotactic polypropylene and form I of isotactic poly(1-butene)[J]. Macromolecules,2011,44:540-549.
[15] NEDOREZOVAA P M,CHAPURINAA A V,KOVAL’CHUKA A A,et al. Copolymerization of propylene with 1-butene and 1-pentene with the isospecific catalytic system rac-Me2Si(4-Ph-2-MeInd)2ZrCl2–MAO[J]. Poly. Sci. Series B,2012,54:1-14.
[16] STAGNARO P,BORAGNO L,LOSIO S,et al. Isoselectivity and steric hindrance of C2-symmetric metallocenes as the keys to control structural and thermal features of ethene/4-methyl-1-pentene copolymers[J]. Macromolecules,2011,44:3712-3722.
[17] ARNOLD M,BORNEMANN S,KOLLER F,et al. Synthesis and characterization of branched polypropenes obtained by metallocene catalysis[J]. Macromol. Chem. Phys.,1998,199:2647-2653.
[18] DERLIN S,KAMINSKY W. Copolymerization of ethylene and propylene with the sterically hindered monomer 3-methyl-1-butene by homogeneous catalysis[J]. Macromolecules,2007,40:4130-4137.
[19] HENSCHKE O,KOLLEL F,ARNOLD M. Polyolefins with high glass transition temperatures[J]. Macromol. Rapid Commun.,1997,18:617-623.
[20] KAMINSKY W,BEULICH I,ARNDT-ROSENAU M. Copolymerization of ethene with cyclic and other sterically hindered olefins[J]. Macromol. Symp.,2001,173:211-225.
[21] de ROSA C,AURIEMMA F. From entropic to enthalpic elasticity:novel thermoplastic elastomers from syndiotactic propylene-ethylene copolymers[J]. Adv. Mater.,2005,17:1503-1507.
[22] de ROSA C,AURIEMMA F,CORRADI M,et al. Mechanical properties and elastic behavior of syndiotactic propene-butene copolymers[J]. Macromolecules,2009,42:4728–4738.
[23] LUO Y J,BALDAMUS J,HOU Z M. Scandium half-metallocene-catalyzed syndiospecific styrene polymerization and styrene-ethylene copolymerization:unprecedented incorporation of syndiotactic styrene-styrene sequences in styrene-ethylene copolymers[J]. J. Am. Chem. Soc.,2004,126:13910- 13911.
[24] NOMURA K,FUKUDA H,APISUK W,et al. Ethylene copolymerization by half-titanocenes containing imidazolin-2-iminatoligands–MAO catalyst systems[J]. J. Mol. Cat. A:Chem.,2012,363/364:501–511.
[25] BENSASON S,MINICK J,MOET A,et al. Classification of homogeneous ethylene-octene copolymers based on comonomer content[J]. Polym. Sci.,Part B:Polym. Phys.,1996,34:1301-1315.
[26] Symyx Technologies Inc. Bridged bi-aromatic ligand catalysts,process for polymerizing and polymers therefrom:US 6897276B2[P]. 2005-05-24.
[27] Exxon Chemical Patent Inc. High temperature olefin polymerization process:US005907021A[P]. 1999-05-25.
[28] Dow Chemical Company. Constrained geometry additionpolymerization catalysts,process for their preparation,precursors therefor,methods of use,and novel polymers therewith:EP 0416815B1[P]. 1997-08-13.
[29] Dow Global Technologies LLC. High temperature solution polymerization process:US8354484B2[P]. 2013-01-15.
[30] NABIKA M,KATAYANA H,WATANABE T,et al. Ansa-cyclopentadienyl-phenoxy titanium (Ⅳ) complexes (PHENICS):synthesis,characterization,and catalytic behavior in olefin polymerization[J]. Organometallics,2009,28:3785-3792.
[31] NOVA Chemicals (International) S A. High temperature solution polymerization process: EP0881233A1[P]. 1998-02-12.
[32] DSM IP Assets BV. Polymerization catalyst comprising an amidine ligand:WO2005/090418A1[P]. 2005-09-29.
[33] SK Innovation Co. Ltd. Ethylene-propylene-diene copolymer production method:EP2377894A2[P]. 2011-10-19.
[34] Equistar Chemicals L P. High temperature solution process for polyolefin manufacture:US6756455B2[P]. 2004-01-29.
[35] XU G X,RUCKENSTEIN E. Ethylene copolymerization with 1-octene using a 2-methylbenz[e]indenyl-based ansamonocyclopentadienylamido complex and methylaluminoxanes catalyst[J]. Macromolecules,1998,31:4724-4729.
[36] CANOA J,KUNZ K. How to synthesize a constrained geometry catalyst (CGC) – A survey[J]. J. Organomet. Chem.,2007,692:4411-4423.
[37] RYABOV A N,VOSKOBOYNIKOV A Z. Constrained geometry complexes of titanium (Ⅳ) and zirconium (Ⅳ) involving cyclopentadienyl fused to thiophene ring[J]. J. Organomet. Chem.,2005,690:4213-4221.
[38] GRANDINI C,CAMURATI I,GUIDOTTI S,et al. Heterocycle-fused indenyl silyl amido dimethyl titanium complexes as catalysts for high molecular weight syndiotactic amorphous polypropylene[J]. Organometallics,2004,23:344-360.
[39] IRWIN L J,REIBENSPIES J H,MILLER S A. A sterically expanded “constrained geometry catalyst” for highly active olefin polymerization and copolymerization:an unyielding comonomer effect[J]. J. Am. Chem. Soc.,2004,126:16716-16717.
[40] CHAI J F,ABBOUD K A,MILLER S A. Sterically expanded CGC catalysts:substituent effects on ethylene and α-olefin polymerization[J]. Dalton Trans.,2013,42:9139-9147.
[41] WU C J,LEE S H,YUN H,et al. Ortho lithiation of tetrahydroquinoline derivatives and its use for the facile construction of polymerization catalysts[J]. Organometallics,2007,26:6685-6687.
[42] WU C J,LEE S H,YU S T,et al. CO2-Mediated ortho-lithiation of N-alkylanilines and its use for the construction of polymerization catalysts[J]. Organometallics,2008,27:3907-3917.
[43] TAO X,WU Q L,HUO H,et al. New titanium (IV) complexes with 2 cyclopentadienylbenzylamido ligands:synthesis,characterization,and catalytic properties for ethylene polymerization and copolymerization with 1-hexene[J]. Organometallics,2013,32:4185-4191.
[44] NOMURA K. Half-titanocenes containing anionic ancillary donor ligands as promising new catalysts for precise olefin polymerization[J]. Dalton Trans.,2009:8811-8823.
[45] NOMURA K,OYA K,KOMATSU T,et al. Effect of the cyclopentadienyl fragment on monomer reactivities and monomer sequence distributions in ethylene/a-olefin copolymerization by a nonbridged (cyclopentadienyl)(aryloxy)titanium(Ⅳ) complex-MAO catalyst system[J]. Macromolecules,2000,33:3187-3189.
[46] NOVA Chemicals (International) S A. Catalysts having a ketimine ligand:US006114481A[P]. 2000-09-05.
[47] KIM T J,KIM S K,KIM B J,et al. Half-metallocene titanium (Ⅳ) phenyl phenoxide for high temperature olefin polymerization:Ortho-substituent effect at ancillary o-phenoxy ligand for enhanced catalytic performance[J]. Macromolecules,2009,42:6932-6943.
[48] NOMURA K. Half-titanocenes containing anionic ancillary donor ligands:effective catalyst precursors for ethylene/styrene copolymerization[J]. Catalysts,2013,3:157-175.
[49] NOMURA K. Syndiospecific styrene polymerization and ethylene/ styrene copolymerization using half-titanocenes:ligand effects and some new mechanistic aspects[J]. Catal. Surv. Asia,2010,14:33-49.
[50] FRAZIER K A,FROESE R D,HE Y Y,et al. Pyridylamido hafnium and zirconium complexes:synthesis,dynamic behavior,and ethylene/1-octene and propylene polymerization reactions[J]. Organometallics,2011,30:3318-3329.
[51] FONTAINE P P,FIGUEROA R,MCCANN S D,et al. Synthesis and scale-up of imino-enamido hafnium and zirconium olefin polymerization catalysts[J]. Organometallics,2013,32,2963-2972.
[52] SZUROMI E,KLOSIN J,ABBOUD K A. Aminotroponiminato hafnium and zirconium complexes:synthesis and ethylene/1-octene copolymerization study[J]. Organometallics,2011,30:4589-4597.
[53] FONTAINE P P,KLOSIN J,MCDOUGAL N T. Hafnium amidoquinoline complexes:highly active olefin polymerization catalysts with ultrahigh molecular weight capacity[J]. Organometallics,2012,31:6244-6251.
[54] Mitsubishi Chemical Corporation. Thermoplastic elastomer composition and method for production same:WO2013/061974A1[P].2013-05-02.
[55] ARRIOLA D J,BOKOTA M,CAMPBELL R E,et al. Penultimate effect in ethylene-styrene copolymerization and the discovery of highly active ethylene-styrene catalysts with increased styrene reactivity[J]. J. Am. Chem. Soc.,2007,129:7065-7076.
[56] NGUYENA T D H,NGUYENA T L T,NOH S K,et al. Bridge length effect of new dinuclear constrained geometry catalysts on controlling the polymerization behaviors of ethylene/styrene copolymerization[J]. Polymer,2011,52:318-325.
[57] TERAO H,IWASHITA A,ISHII S,et al. Ethylene/norbornene copolymerization behavior of bis(phenoxy-imine)Ti complexes combined with MAO[J]. Macromolecules,2009,42:4359-4361.
[58] WANG C,SUN X L,GUO Y H,et al. Novel titanium catalysts bearing an[O,N,S] tridentate ligand for ethylene homo- and copolymerization[J]. Macromol. Rapid Commun.,2005,26:1609-1614.
[59] NOMURA K,FUKUDA H,KATAO S,et al. Olefin polymerization by half-titanocenes containing η2-pyrazolatoligands-MAO catalyst systems[J]. Macromolecules,2011,44:1986-1998.
[60] TANG X Y,WANG Y X,LI B X,et al. Highly efficient ethylene/norbornene copolymerization by o-di(phenyl) phosphanylphenolate- based half- titanocene complexes[J]. Polym. Sci.,Part A:Polym. Chem.,2013,51:1585-1594.
[61] KHAN F Z,KAKINUKI K,NOMURA K. Copolymerization of ethylene with tert-butylethylene using nonbridged half-titanocene-cocatalyst systems[J]. Macromolecules,2009,42:3767-3773.
[62] NOMURA K,ITAGAKI K. Efficient incorporation of vinylcylohexane in ethylene/vinylcyclohexane copolymerization catalyzed by nonbridged half- titanocenes[J]. Macromolecules,2005,81:8121-8123.
[63] WANG W,FUJIKI M,NOMURA K. Copolymerization of ethylene with cyclohexene (CHE) catalyzed by nonbridged half-titanocenes containing aryloxo ligand:notable effect of both cyclopentadienyl and anionic donor ligand for efficient che incorporation[J]. J. Am. Chem. Soc.,2005,127:4582- 4583.
[64] HEUER B,KAMINSKY W. Alternating ethene/propene copolymers by C1-symmetric metallocene/mao catalysts[J]. Macromolecules,2005,38:3054-3059.
[65] UOZUMI T,AHN C H,TOMISAKA M,et al. Synthesis of ethylene-a-olefin alternating copolymers with Et(1-Ind)(9-Flu)ZrCl2-MAO catalyst system[J]. Macromol. Chem. Phys.,2000,201:1748-1752.
[66] KAMINSKY W,PIEL C. Tailoring polyolefins by metallocene catalysis:kinetic and mechanistic aspects[J]. J. Mol. Cat. A:Chem.,2004,213:15–19.
[67] EWEN J A,ELDER M J,JONES R L,et al. Chiral ansa metallocenes with Cp ring-fused to thiophenes and pyrroles:syntheses,crystal structures,and isotactic polypropylene catalysts[J]. J. Am. Chem. Soc.,2001,123:4763-4773.
[68] SCHOBEL A. Differences of zirconocenes and hafnocenes from low isotactic,elastic- to high isotacticity,high melting polypropylene[D]. Munchen:Technische Universitat Munchen,2012.
[69] MILLER S A,BERCAW J E. Mechanism of isotactic polypropylene formation with C1-symmetricmetallocene catalysts[J]. Organometallics,2006,25:3576-3592.
[70] BADER M,MARQUET N,KIRILLOV E,et al. Old and new C1symmetric group 4 metallocenes {(R1R2C)-(R2′R3′R6′R7′-Flu) (3-R3-5- R4-C5H2)}ZrCl2:from highly isotactic polypropylenes to vinyl end-capped isotactic-enriched oligomers[J]. Organometallics,2012,31:8375-8387.
[71] Dow Global Technologies INC. Isotactic propylene copolymer fibers,their preparation and use:US 6906160B2[P]. 2005-06-14.
[72] IRWIN L J,MILLER S A. Unprecedented syndioselectivity and syndiotactic polyolefin melting temperature:polypropylene and poly(4-methyl-1-pentene) from a highly active,sterically expanded η1-fluorenyl-η1-amido zirconium complex[J]. J. Am. Chem. Soc.,2005,127:9972-9973.
[73] Fina Technology Inc. Syndiotactic polypropylene and method of preparing same:US2008/0097052A1[P]. 2008-04-24.
[74] Basell Polyolefin GmbH. BiIndenyl zirconium complexes for use in polymerization of olefins:US2006/0252637A1[P]. 2006-11-09.
[75] NIFANT’EV L E,IVCHENKOV P V,BAGROV V V,et al. Asymmetric ansa-zirconocenes containing a 2-methyl-4-aryltetrahydroindacene fragment:synthesis,structure,and catalytic activity in propylene polymerization and copolymerization[J]. Organometallics,2011,30:5744-5752.
[76] NIFANT’EV L E,IVCHENKOV P V,BAGROV V V,et al. Novel effective racemoselective method for the synthesis of ansazirconocenes and its use for the preparation of C2-symmetriccomplexes based on 2-methyl-4-aryltetrahydro(s)indacene as catalysts for isotactic propylene polymerization and ethylene/propylene copolymerization[J]. Organometallics,2012,31:4340-4348.
[77] RESCONI L,CAMURATI I,MALIZIA F. Metallocene catalysts for 1-butene polymerization[J]. Macromol. Chem. Phys.,2006,207:2257-2279.
·技术信息·
Progress in advanced single site catalysts for preparing all series polyolefin materials
MAIMAITIMING Aizezi
(State Key Laboratory of Chemical Engineering,College of Chemical & Biological Engineering,Zhejiang University,Hangzhou 310027,Zhejiang,China)
Abstract:The development of advanced single site catalysts provided us tremendous opportunities for designing and tailoring the molecular chain structure of polyolefins. However,designing,preparing and optimizing of high temperature stable multi-functional catalysts became core issue to develop high performance and high value added polyolefin products. Firstly,this paper introduced all types of series polyolefin materials and the correlation of melting properties and material behaviors with comonomer content. Secondly,we mainly reviewed:①high temperature stable ethylene/α-olefin copolymerization catalysts,covering modified CGC-titanocenes/half-titanocenes/non-metallocene hafnium; ②half-titanocene and non-metallocene titanium catalysts for ethylene/(styrene,norborene) ser-copolymerization; ③modified C2/C1-symmetric zirconium and high temperature stable non-metallocene hafnium catalysts for stereo-regular propylene homopolymerization and isotactic propylene/ethylene ser-copolymerization; ④heterocycle-containing C2/C1-symmetric zirconocenes for isotactic 1-butene bulk solution polymerization. Meanwhile,more emphases were given to the polymerization behaviors and the tailored chain structures of the corresponding products of these catalysts. At last,it was suggested that future research should be focused on optimizing isotacticbook=111,ebook=118non-metallocene hafniums,hetero-cycle/atom containing C2/C1- symmetric zirconocenes and syndiotactic C1-symmetric zirconocenes.
Key words:metallocenes; non-metallocenes; catalyst; polyolefins; preparation; high throughput catalyst screening (HTCS); chain structure
基金项目:国家重点基础研究发展计划项目子课题(2011CB606001)及国家自然科学基金重点项目(20936006)。
收稿日期:2015-04-27;修改稿日期:2015-07-01。
DOI:10.16085/j.issn.1000-6613.2016.01.015
中图分类号:TQ 325.1
文献标志码:A
文章编号:1000–6613(2016)01–0110–15
