新型四阳离子型1,2,3-三唑鎓环蕃的合成
2016-03-01段群鹏
段群鹏, 卢 奎
(1. 河南工程学院 材料与化学工程学院,河南 郑州 451191;
2. 河南工业大学 化学化工学院,河南 郑州 450052)
·快递论文·
新型四阳离子型1,2,3-三唑鎓环蕃的合成
段群鹏1*, 卢奎1,2
(1. 河南工程学院 材料与化学工程学院,河南 郑州451191;
2. 河南工业大学 化学化工学院,河南 郑州450052)
摘要:以间苯二酚为原料,经5步反应制得中间体4,4′-[4,6-双(己氧基)-1,3亚苯基]双{1-[4-(叠氮甲基)苯基]-1H-1,2,3-三氮唑}(6); 6与1,5-二乙炔基-1,4-二(己氧基)苯在高度稀释的条件下经Click反应环合制得1,2,3-三唑环蕃(7); 7与三甲基氧鎓四氟硼酸盐反应合成了全甲基化的新型1,2,3-三唑鎓环蕃,其结构经1H NMR,13C NMR和HR-ESI-MS表征。
关键词:四阳离子; 1,2,3-三唑鎓; 环蕃; 合成
环蕃是一类以平面芳环为构筑基元,通过脂肪链连接而形成的环状分子[1]。环蕃不仅兼备冠(穴)醚、环糊精和多齿配体的特性和结构优势,还具有合成方法与结构修饰的灵活多样性,能更好的发挥氢键、疏水作用、静电作用、π-π、阳离子-π的协同效应,故在主-客体化学、超分子化学、模拟酶、仿生化学、分子自组装及材料科学领域受到广泛关注[2]。
三唑蕃是一类由一个或多个三唑环通过各种分子模块连接而成的新型环蕃[3]。三唑蕃因其三唑环的π-π共轭体系,环上2个碳原子上的电子云密度较低,5-位碳原子上的氢原子可与各种阴离子形成氢键,这种非共价键相互作用具有方向性,与多种阴离子亲和力较好[4]。更为重要的是,三唑环可与含有氢原子的苯环、吡咯等芳香环形成环状空腔结构,并包结阴离子形成超分子络合物。此外,三唑环作为阴离子受体,对卤素离子和焦磷酸根离子等阴离子均有较好的识别作用[5-7]。三唑蕃中三唑环上的氮原子易季铵化形成三唑鎓蕃。三唑鎓蕃因三唑鎓提供的正电荷,可以更好的适应客体的结构与电学特性,从而成为一种良好的主体分子[8-9]。但在环蕃主体中引入三唑鎓越多,其合成和分离的难度越大。因此近年来鲜有关于这种包含四个三唑鎓组分的大环蕃的报道。
基于此,本文以间苯二酚为原料,经5步反应制得中间体4,4′-[4,6-双(己氧基)-1,3-亚苯基]双{1-[4-(叠氮甲基)苯基]-1H-1,2,3-三氮唑]}(6); 6与1,5-二乙炔基-1,4-二(己氧基)苯在高度稀释的条件下经Click反应环合制得1,2,3-三唑环蕃(7); 7与三甲基氧鎓四氟硼酸盐反应合成了全甲基化的新型1,2,3-三唑鎓环蕃(8, Scheme 1),其结构经1H NMR,13C NMR和LR-ESI-MS表征。
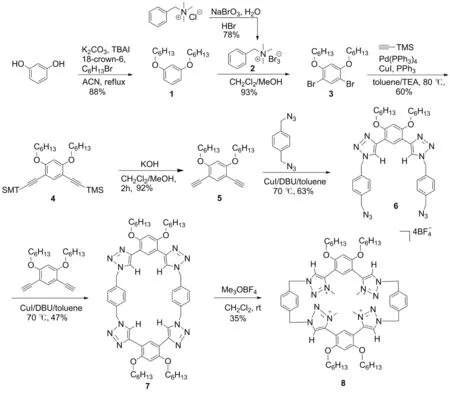
Scheme 1
1实验部分
1.1仪器与试剂
X-4型熔点仪(温度未校正);Bruker AVANCE DMX 400 MHz型核磁共振仪(CDCl3或DMSO-d6为溶剂,TMS为内标);Finnigan Mat TSQ 7000 型质谱仪。
1,4-二叠氮甲基苯按文献[10]方法合成;1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU),上海阿拉丁生化科技股份有限公司;其余所用试剂均为分析纯,使用前按标准方法纯化。
1.2合成
(1) 1,3-二(己氧基)苯(1)的合成[11]
氩气保护下,在三口烧瓶中依次加入间苯二酚16.6 g(150.0 mmol), K2CO362.2 g, 18-冠-6 1.98 g,四丁基碘化铵(TBAI)5.54 g和脱气乙腈200 mL,搅拌使其溶解;加入溴己烷42.6 mL(300 mmol),回流反应过夜(TLC检测)。冷却至室温,过滤,滤液旋蒸浓缩后经硅胶柱层析[洗脱剂:A=V(石油醚) ∶V(乙酸乙酯)=8 ∶1]纯化得红色油状液体1 36.7 g,收率88%;1H NMRδ: 7.15(t,J=8.1 Hz, 1H), 6.48(m, 3H), 3.93(t,J=6.6 Hz, 4H), 1.81~1.72(m, 4H), 1.45~1.26(m, 12H), 0.90(t,J=6.6 Hz, 6H)。
(2) 苄基三甲基三溴化铵(2)的合成[12]
在单口烧瓶中依次加入苄基三甲基氯化铵34.1 g(180.0 mmol),溴酸钠13.6 g (90.0 mmol)和水300 mL,搅拌使其溶解;加入40%氢溴酸73.3 mL,于室温反应1 h(TLC检测)。抽滤,滤饼用二氯甲烷(4×150 mL)洗涤,合并滤液和洗液,用无水硫酸镁干燥;过滤,滤液真空浓缩后用混合溶剂[V(二氯甲烷) ∶V(乙醚)=10 ∶1]重结晶得橙色晶体2 55.4 g,收率79%, m.p.100~102 ℃。
(3) 1,5-二溴-2,4-二(己氧基)苯(3)的合成[12]
在单口烧瓶中加入1 36.7 g(132.0 mmol), 2 53.02 g(136.0 mmol),二氯甲烷500 mL和甲醇200 mL,搅拌下于室温反应10 h(TLC检测)。旋蒸除溶,残余物加水200 mL,用乙醚萃取,合并有机相,用无水硫酸镁干燥;过滤,滤液经真空浓缩后经硅胶柱层析(洗脱剂:A= 8 ∶1)纯化得橙色固体3 26.14 g,收率93%;1H NMRδ: 7.64(s, 1H), 6.46(s, 1H), 3.99(t,J=6.4 Hz, 4H), 1.87~1.78(m, 4H), 1.56~1.48(m, 4H), 1.36~1.26(m, 8H), 0.91(t,J=6.3 Hz, 6H)。
(4) 2,4-二(己氧基)-1,5-二(三甲基硅-乙炔基)苯(4)的合成[11]
氩气保护下,在三口烧瓶中依次加入3 6.0 g(13.76 mmol),碘化亚铜210 mg,四(三苯基膦)钯1.27 mg,三苯基膦362 mg,脱气甲苯100 mL和干燥三乙胺70 mL,搅拌使其溶解;加入三甲基乙炔基硅5.9 mL(41.28 mmol),搅拌下于80 ℃反应3 d(TLC检测)。冷却至室温,过滤,滤液浓缩,残余物用二氯甲烷溶解,依次用水和饱和食盐水洗涤,无水硫酸镁干燥;过滤,滤液浓缩后经硅胶柱层析(洗脱剂:石油醚)纯化得淡黄色固体4 3.85 g,收率60%;1H NMRδ: 7.50(s, 1H), 6.30(s, 1H), 3.99(t,J=6.3 Hz, 4H), 1.87~1.78(m, 5H), 1.55~1.48(m, 8H), 1.37~1.31(m, 12H), 0.93~0.83(m, 6H), 0.23(s, 18H)。
(5) 1,5-二乙炔基-2,4-二(己氧基)苯(5)的合成[13]
在单口烧瓶中依次加入4 2.29 g(4.87 mmol),脱气甲醇40 mL, 0.1 mol·L-1氢氧化钾溶液1.4 mL和二氯甲烷5 mL,搅拌下于室温反应2 h(TLC检测)。旋蒸除溶,残余物用二氯甲烷100 mL溶解,用水(2×100 mL)洗涤,无水硫酸镁干燥;过滤,滤液浓缩得淡黄色固体5 1.46 g,收率92%;1H NMRδ: 7.53(s, 1H), 6.37(s, 1H), 4.03(t,J=6.5 Hz, 4H), 3.16(s, 2H), 1.89~1.80(m, 4H), 1.55~1.45(m, 4H), 1.35(m, 8H), 0.93~0.83(m, 6H)。
(6) 6的合成[14]
氩气保护下,在三口烧瓶中依次加入1,4-二叠氮甲基苯376.0 mg(2.0 mmol),碘化亚铜90.0 mg, DBU 0.6 mL和脱气甲苯20 mL,搅拌下于70 ℃缓慢滴加5 163.0 mg(0.5 mmol)的甲苯(30 mL)溶液,滴毕(12 h内),反应2 h(TLC检测)。冷却至室温,旋蒸除溶,残余物经硅胶柱层析[洗脱剂:B=V(二氯甲烷) ∶V(甲醇) =100 ∶1]纯化得黑色晶状固体6 220 mg,收率63%, m.p.109~111 ℃;1H NMRδ: 9.13(s, 1H), 7.87(s, 2H), 7.33(s, 8H), 6.49(s, 1H), 5.59(s, 4H), 4.35(s, 4H), 4.03(t,J=6.4 Hz, 4H), 1.82~1.70(m, 4H), 1.44~1.26(m, 12H), 0.90(t,J=6.8 Hz, 6H);13C NMRδ: 156.2, 143.9, 135.9, 135.4, 128.9, 128.5, 127.4, 122.2, 112.5, 96.4, 68.6, 54.4, 53.7, 31.6, 29.3, 25.9, 22.7, 14.2; LR-ESI-MSm/z: 703.35{[M+H]+}, 725.30{[M+Na]+}。
(7) 7的合成[14]
氩气保护下,在三口烧瓶中依次加入DBU 1.6 mL,脱气甲苯200 mL和碘化亚铜52 mg,搅拌下用恒流注射泵于70 ℃滴加5 89.0 mg(0.3 mmol)和6 190.0 mg(0.3 mmol)的甲苯(50 mL)溶液,滴毕(24 h内),反应4 h(TLC检测)。冷却至室温,旋蒸除溶,残余物经硅胶柱层析(洗脱剂:B=60 ∶1)纯化得白色固体7 130 mg,收率47%, m.p.>250 ℃;1H NMRδ: 8.40(s, 2H), 8.17(s, 4H), 7.24(s, 8H), 6.71(s, 2H), 5.61(s, 8H), 4.10(t,J=5.4 Hz, 8H), 1.86~1.77(m, 8H), 1.41~1.28(m, 24H), 0.84(t,J=6.6 Hz, 12H);13C NMRδ: 155.4, 142.4, 136.6, 128.2, 126.7, 122.6, 111.8, 97.3, 68.3, 52.4, 30.9, 28.4, 25.2, 22.1, 13.8; LR-ESI-MSm/z: 1 029.50{[M+H]+}。
(8) 8的合成[9,15]
氩气保护下,在两口烧瓶中加入7 70 mg(0.07 mmol),三甲基氧鎓四氟硼酸49.7 mg(0.34 mmol)和干燥二氯甲烷20 mL,搅拌下于室温反应3 d(TLC检测)。滴加甲醇10滴淬灭反应,旋蒸除溶,残余物经硅胶柱层析(洗脱剂:B=20 ∶1)纯化得淡黄色固体8 35 mg,收率35%;1H NMRδ: 8.45(s, 4H), 7.55(s, 8H), 7.39(s, 2H), 6.63(s, 2H), 5.75(s, 8H), 4.12~4.09(m, 20H), 1.77~1.72(m, 8H), 1.30(m, 26H), 0.86~0.84(m, 14H);13C NMRδ: 154.7, 137.1, 133.2, 127.2, 126.6, 123.0, 114.3, 99.6, 69.0, 62.0, 33.2, 31.8, 29.6, 25.6, 22.7, 14.1; HR-ESI-MSm/z: Calcd for C64H88N12O4B2F8{[M-2BF4]2+} 631.54, found 631.50; Calcd for C64H88N12O4BF4{[M-3BF4]3+} 392.09, found 392.00。
2结果与讨论
合成.5时,文献[16]方法使用K2CO3,四丁基氟化铵(TBAF)或KOH均可脱除炔基保护基TMS。本实验中单独使用K2CO3并未得到5,换用TBAF或KOH则成功脱去TMS。实验中最终采用KOH脱除TMS,反应在2 h内结束,5纯度较高,无需纯化。
合成6时,由于叠氮化物光分解或热裂解容易放出氮气生成氮烯,6经柱层析纯化时可能会因室温较高及柱层析时使用CH2Cl2(CH2Cl2和硅胶的吸附均为放热过程)导致6部分分解放出氮气形成氮烯结构,故6纯化时需使用快速柱层析分离。
合成7时采用了高度稀释法。该方法是在大量溶剂中,通过控制反应物的滴加速度,减少副反应发生,从而提高收率。实验中使用恒流注射泵精确控制反应物滴加速度,使收率由5%(恒压滴液漏斗)提高至47%。
合成8的反应涉及三氮唑甲基化反应。最初是以MeI作甲基化试剂,但反应较长时间后TLC仍可检测到7, 其原因可能是7的大环具有较强的空间位阻,导致该反应需要较高的能量和较好的甲基化试剂。因此停止反应,旋蒸除去MeI,加入米尔文盐——三甲基氧鎓四氟硼酸作甲基化试剂[9,15],继续反应3 d, TLC检测反应完全。
参考文献
[1]宗婷,孙小强,席海涛. 新型含硫代乙酸酯缺电子环蕃的合成[J].合成化学,2014,22(6):778-780.
[2]席海涛,孙小强,孟启,等.缺电子联吡啶环蕃的研究[J].化学进展,2008, 20(1):88-90.
[3]Mohan A, Sankararaman S. 1,2,3-Triazolophanes—cyclophanes with an array of molecular structures and supramolecular architectures [J].Isr J Chem,2012,52(1-2):92-104.
[4]常娟娟,王艳,张慧珍,等. 三唑类超分子化学与药物研究新进展[J].高等学校化学学报,2011,32(9):1970-1985.
[5]Li Y J, Flood A H. Pure C-H hydrogen bonding to chloride ions:A preorganized and rigid macrocyclic receptor [J].Angew Chem Int Ed,2008,47(14):2649-2652.
[6]Li Y J, Flood A H. Strong,size-selective,and electronically tunable C-H┉halide binding with steric control over aggregation from synthetically modular,shape-persistent [34]triazolophanes [J].J Am Chem Soc,2008,130(36):12111-12222.
[7]Sessler J L, Cai J, Gong H Y,etal. A pyrrolyl-based triazolophane:A macrocyclic receptor with CH and NH donor groups that exhibits a preference for pyrophosphate anions [J].J Am Chem Soc,2010,132(40):14058-14060.
[8]White N G, Carvalho S, Félix V,etal. Anion binding in aqueous media by a tetra-triazolium macrocycle [J].Org Biomol Chem,2012,10(34):6951-6959.
[9]Cai J J, Hay B P, Young N J,etal. A pyrrole-based triazolium-phane with NH and cationic CH donor groups as a receptor for tetrahedral oxyanions that functions in polar media [J].Chem Sci,2013,4:1560-1567.
[10]Demko ZP, Sharpless K B. A click chemistry approach to tetrazoles by Huisgen 1,3-dipolar cycloaddition:Synthesis of 5-sulfonyl tetrazoles from azides and sulfonyl cyanides [J].Angew Chem Int Ed,2002,41(12):2110-2113.
[11]Zornik D, Meudtner, R K, Malah T E,etal. Designing structural motifs for clickamers:Exploiting the 1,2,3-triazole moiety to generate conformationally restricted molecular architectures [J].Chem Eur J,2011,17(5):1473-1484.
[12]Kajigaeshi S, Kakinami T, Tokiyama H,etal. Synthesis of dibromoacetyl derivatives by use of benzyltrimethylammonium tribromide [J].Bull Chem Soc Jpn,1987,60(7):2667-2668.
[13]Neenan TX, Whitesides G M. Synthesis of high carbon materials from acetylenic precursors:Preparation of aromatic monomers bearing multiple ethynyl groups [J].J Org Chem,1988,53(11):2489-2496.
[14]Duan QP, Xia W, Lin C,etal. Two new triazolophanes:Synthesis,structures,self-assembling,and anion complexation properties [J].Tetrahedron Lett,2015,56(26):4002-4006.
[15]Mullen KM, Mercurio J, Serpell C J,etal. Exploiting the 1,2,3-triazolium motif in anion-templated formation of a bromide-selective rotaxane host assembly [J].Angew Chem Int Ed,2009,48:4781-4784.
[16]Jenny NM, Mayor M, Eaton T R. Phenyl-acetylene bond assembly:A powerful tool for the construction of nanoscale architectures [J].Eur J Org Chem,2011,(26):4965-4983.
Synthesis of A Novel
Tetracationic 1,2,3-Triazolium Cyclophane
DUAN Qun-peng1*,LU Kui1,2
(1. College of Material and Chemistry Engineering, Henan Institute of Engineering, Zhengzhou 451191, China
2. School of Chemistry and Chemical Engineering, Henan University of Technology, Zhengzhou 450052, China)
Abstract:An intermediate, 4,4′-[4,6-bis(hexyloxy)-1,3-phenylene]bis{1-[4-(azidomethyl)benzyl]- 1H-1,2,3-triazole}(6), was prepared by a five-step reaction from resorcinol. 1,2,3-Triazolophane(7) was obtained by Click reaction of 6 with 1,5-diethynyl-2,4-bis(hexyloxy)benzene under pseudo-high dilution conditions. A novel tetramethylated 1,2,3-triazolium-phane was synthesized by the reaction of trimethyloxonium tetrafluoroborate with 7. The structure was characterized by1H NMR,13C NMR and HR-ESI-MS.
Keywords:tetracationic; 1,2,3-triazolium; cyclophane; synthesis
作者简介:段群鹏(1981-),男,汉族,河南鹿邑人,博士,讲师,主要从事大环超分子的研究。 E-mail: dqp117@163.com
基金项目:国家自然科学基金资助项目(21402040, 21172054)
收稿日期:2015-11-05;
修订日期:2015-12-22
中图分类号:O621.3; O626.26
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.02.15371
