基于特征加权的蛋白质交互识别
2016-02-23吴红梅
吴红梅,牛 耘
(南京航空航天大学 计算机科学与技术学院,江苏 南京 210016)
基于特征加权的蛋白质交互识别
吴红梅,牛 耘
(南京航空航天大学 计算机科学与技术学院,江苏 南京 210016)
在以单词为特征的模型中,如果特征单词在不同类别中的使用情况存在明显差异,那么它对分类有着很重要的影响。因此文中基于大规模语料库,研究不同的特征加权方法对PPI识别的影响。首先,通过搜索医学文献数据库建立蛋白质对的签名档,以单词作为描述蛋白质对关系的特征,构建向量空间模型;然后,选择不同的加权方法描述单词重要性;最后,以K近邻和SVM分类方法构建分类器判断蛋白质对是否存在交互关系。实验结果表明,根据特征向量单词的重要性进行加权,PPI识别精确度、召回率和准确率有了明显的提高。
蛋白质交互;大规模语料;特征加权;K近邻;支持向量机
0 引 言
蛋白质是组成细胞最重要的成分,是生命的物质基础,是生命活动的主要承担者。蛋白质交互(Protein-ProteinInteraction,PPI)是生物学研究的重要内容,也是解决大量医学难题的关键信息。因此,为了构建蛋白质交互网络,生物医学领域的专家从医学文献中手工整理信息以统一的格式录入数据库,构建了HPRD[1]、IntAct[2]、MINT[3]等数据库。
然而,随着生物医学的发展,越来越多的蛋白质交互关系被发现,记载这些蛋白质交互信息的医学文献也随之急剧增多,手工收集PPI信息的方式也难以满足需求。为了帮助生物医学领域的专家从文献中获取有效的信息,基于自然语言处理的蛋白质交互识别已成为一项重要的研究内容。
1 相关工作
目前,常用于从医学文献中识别PPI信息的技术主要包括:基于同现的方法[4]、基于规则的方法[5]和基于机器学习的方法[6-7]。基于同现的方法通过统计两个蛋白质的共现次数来判断蛋白质对之间的关系,这种方法只能抽取频繁出现的模式,识别结果召回率较高而精确度较低[8]。基于规则的方法通过建立一些模式规则来匹配可能出现的PPI关系,虽然提高了精确度但召回率较低。然而,由于PPI关系模式的多样性,预定义的规则不可能包含所有的PPI关系模式,并且手工建立规则需要巨大的开销[9]。例如,Fundel等[10]提出的规则是基于句子句法规则结构中的依赖关系。Temkin等[11]利用带语法产生规则的句子分析器来识别PPI。这些系统着眼于分析整个句子的句法特点,充分揭示句中成分之间的关系,能够获得更高的准确率,但需要更高的计算能力和时间复杂度。
近年来,越来越多的PPI识别技术采用基于机器学习的方法,主要包括两大类:基于特征的方法和基于核函数的方法。基于特征的方法主要是从标注有交互关系的蛋白质对的句子中抽取重要特征,例如词汇、语法和语义特征建立模型,进而判断蛋白质对之间是否存在交互关系[12-13]。基于核函数的方法通过对句子结构的深入分析来构建核函数。HausslerD[14]提出了针对离散结构的卷积核;LodhiH等[15]将特征空间特定长度词语子序列的内积作为核函数的计算方式,提出了字符串核;BunescuRC等[16]提出了最短依赖路径核,将句子以树的形式表示,用两个实体之间的最短路径表示实体之间的关系。
然而,目前的机器学习方法主要以单个句子为依据,从句子的句法、语法以及依赖关系等方面进行研究。这些方法能很好地从句子层面对蛋白质交互关系进行描述及判断,但是这种以句子为依据的判断方法也存在着局限性。由于语法的复杂性和交互关系描述的间接性,仅仅依赖单个句子中的信息进行交互关系分析,往往难以得到准确的判断。
因此针对以上问题,文献[17-18]提出了基于大规模语料库的PPI自动识别方法,将PPI自动识别问题转化为文本分类问题,能够更充分利用文本的上下文信息挖掘更多蛋白质对交互识别的影响因素。然而,这个工作对所选择特征单词的重要性研究不足,因此文中研究不同的特征单词重要性计算方法,通过向量加权方法研究特征单词加权对蛋白质对识别的影响。实验结果表明,根据特征单词进行加权后明显提高了PPI识别精确度、召回率和准确率。
2 基于特征加权的PPI识别
有交互关系的描述常用到一些单词,而这些单词在无交互关系的蛋白质对的关系描述中却很少出现,例如,interact、bind。因此,加强这部分单词的作用有助于PPI识别。文中以特征加权的方法,着重研究特征单词权重对PPI识别的影响,具体步骤如下:
首先,从医学文献中收集包含目标蛋白质对的句子,以对应的句子集合作为该目标蛋白质对的签名档;然后,从签名档中提取特征,采取一定策略评估特征重要性,对特征加权,构建向量空间模型;最后,采用KNN和SVM这两种分类算法对蛋白质对进行分类,判断蛋白质对是否存在交互关系。
2.1 获取签名档
生物医学文本数据库PubMed[19]是建立蛋白质交互网络的重要信息来源。现有的PPI识别工作都是建立在对PubMed一个子集上的分析。PubMed数据库由美国国立医学图书馆建立,收录了全球70多个国家及地区出版的3 400余种生物医学期刊上所发表的论文,已收录超过2 100万篇生物医学文献,提供生物医学方面论文以及摘要的搜索。但是,PubMed数据库未提供直接搜索句子的接口,所以文中分以下两步来获取包含目标蛋白质对的句子:
(1)在PubMed数据库中获取同时含有目标蛋白质protein1和protein2的文献摘要;
(2)在第一步得到的文献摘要中找出同时包含protein1和protein2的句子。
因此,每个蛋白质对都会有一个句子集合与之对应,形成蛋白质对的签名档。在建好签名档之后,就可以利用上下文信息分析蛋白质对是否存在交互关系。
2.2 向量空间模型—特征提取
从签名档中提取特征,采取一定策略评估特征的重要性,对特征进行加权处理。文献[17]中选择了单词、短语结构特征和依赖关系作为特征,实验结果表明只以单词为特征的识别结果较好。
因此,文中将签名档中所有的句子去除停止词、单字符单词和数字,选择至少在25篇签名档中出现的单词作为特征。最终得到了4 867个特征,用这些特征单词标注蛋白质,构建向量空间模型。
2.3 向量空间模型—特征加权
在蛋白质对签名档中,常用于描述有交互关系的单词较少用于描述无交互关系。因此,通过特征加权的方法,着重研究这部分单词的重要性。选择了信息检索(Information Retrieval)和情感分析(Sentiment Analysis)中的权重计算方法,并设计了新的加权方法,研究特征加权对PPI识别的影响,具体见表1。

表1 权重公式
注:N表示蛋白质对总数;N+表示有交互关系蛋白质对数;N-表示无交互关系蛋白质对数;a表示出现特征i的有交互关系蛋白质对的签名档数;c表示出现特征i的无交互关系蛋白质对的签名档数。
表1中,tp(termpresence)表示二值权重(0/1),即特征单词出现权值为1,不出现权值为0。实验中,以tp权重公式的实验结果为基准。idf(inversedocumentfrequency)是信息检索中较为常见的权重公式,是一个词语普遍重要性的度量。dsidf和dbidf是PaltoglouG[21]等对idf的一种改进。
在这两个公式中,当有交互蛋白质对和无交互蛋白质对的签名档的数量相近时,而含有某个特征单词的有交互蛋白质对的签名档数多于无交互蛋白质对签名档数,那么这个特征单词的权重较大。这两种权值计算公式比较适合特征单词在不同类别文档中分布差异很大的语料集上使用。
在特征单词分布分析过程中发现,有交互蛋白质对和无交互关系蛋白质对的签名档数量相近。而对一些特征单词而言,包含它的有交互关系蛋白质对的签名档的数量是无交互关系签名档数量的数十倍。笔者认为这些单词对于识别有交互蛋白质很重要,因此,提出了power这一权重公式,着重研究特征单词对有交互关系蛋白质对的识别的影响。根据公式power,若含有某个特征单词的有交互关系的蛋白质对的签名档数较多,无交互关系的蛋白质对的签名档数较少,则它的权重较大。
2.4 分类算法
文中采用KNN和SVM这两种分类算法构建分类器。基于相似性的KNN分类器中,以余弦相似度作为衡量标准。
2.4.1K近邻分类
(1)
余弦值越大,蛋白质对相似度就越高;反之,蛋白质对相似度越低。
得到蛋白质对实例的相似性之后,基于相似性采用K近邻分类(KNN)算法对蛋白质对进行分类。查询训练数据中与目标蛋白质对最相似的K个蛋白质对实例。这K个实例中哪种类别的实例多,就将目标蛋白质对分为哪一类。在此算法中,若多个实例与目标蛋白质对的距离一样,则这个实例类别取这些实例中占多数的类别。
2.4.2SVM分类
SVM已被大量实验证实为一种非常有效的分类算法,是基于机器学习的蛋白质交互关系识别所采用的重要分类模型。文中采用LIBSVM[22]建立蛋白质交互识别的分类器。
3 实 验
3.1 实验数据及设置
将有交互关系的蛋白质对作为正类样例,无交互关系的蛋白质对作为负类样例。正类蛋白质对来源于由专家手工收集信息建立的PPI数据库HPRD,从中抽取在PubMed数据库中存在的蛋白质对作为有交互关系的蛋白质对训练集,共1 420对。而对于负类,文中根据HPRD中包含的蛋白质采用随机组合的方法产生负类蛋白质对(删除HPRD已包含的蛋白质对),最后只保留那些被PubMed数据库中文献记载的蛋白质对作为无交互蛋白质对的训练集,共有1 353对。因此,实验数据集中共包含2 773对蛋白质对。
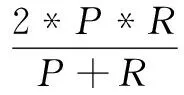
SVM分类算法识别过程中,采用五折交叉验证,将正类蛋白质对和负类蛋白质对平均分成五份,分别从正类和负类中取出一份组合作为测试集,其余四份组合作为训练集。KNN算法识别过程中,采用留一交叉验证法(leave-one-out)进行测试,即将一个蛋白质对作为测试样例,其余2 772对作为训练样例。在以KNN算法为分类算法的实验中,tp在K值为7时取得了最好的结果。所以,在其余的权重公式识别过程中将K值设置为7,将它们的识别结果与tp的识别结果进行比较。
3.2 实验结果及分析
分类结果见表2和表3。
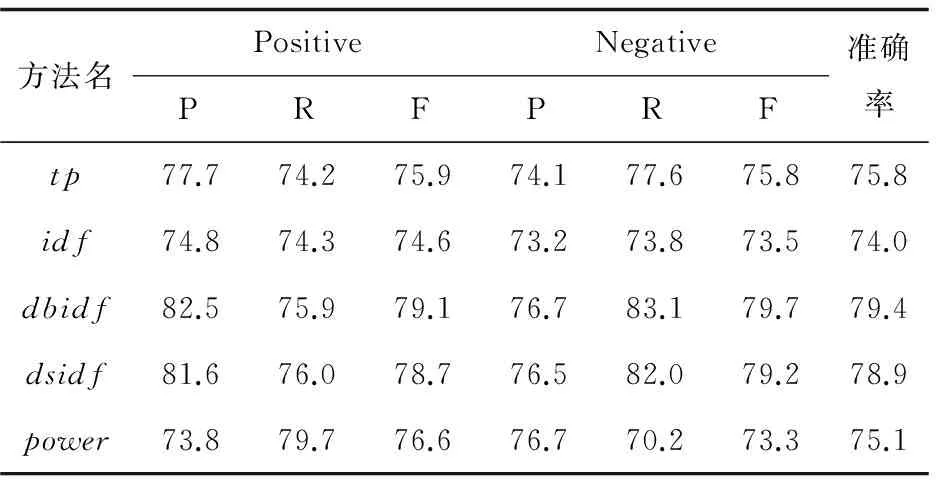
表2 KNN分类结果 %
从表2可以发现,以idf为权重公式的PPI识别结果与tp相比未有提升。以dbidf和dsidf这两种方法为权重公式的PPI识别结果提升较明显,正类、负类的F值和准确率有近3%~5%的提升。笔者设计的权重公式取得了最高的正类的召回率,正类的召回率比tp提升了5.5%,正类F值也有一定的提升。采用dbidf、dsidf和power这三种加权方法的KNN分类算法,蛋白质交互关系的识别结果都有了一定的提升。这说明,特征单词在正类和负类蛋白质对签名档有很大的差别,对蛋白质对交互关系识别起到很大的帮助。
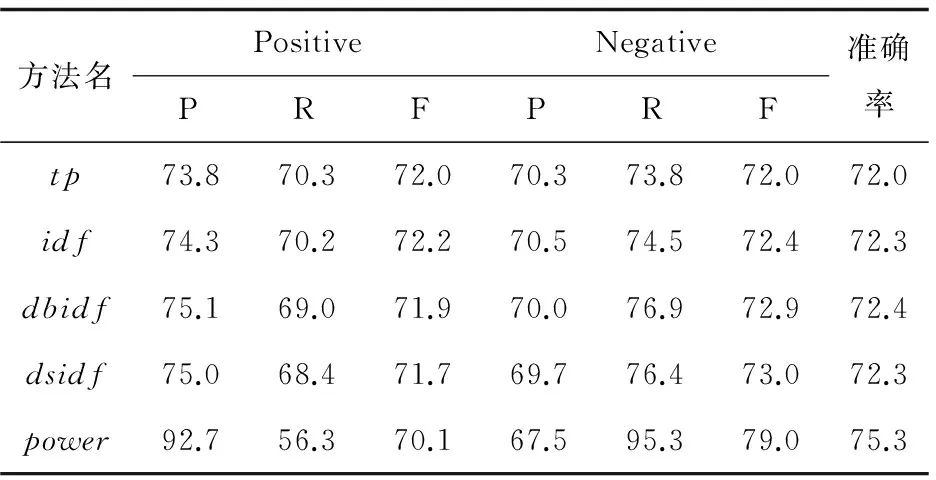
表3 SVM分类结果 %
从表3中发现,idf、dbidf和dsidf这三种方法的识别结果较tp没有明显提升。而笔者设计的权重公式正类精确度达到了92.7%,比tp提高了18.9%。负类蛋白质对的F值比tp提高了7%,并且总体分类准确率较tp提高了3.3%。这说明,采用power为权重公式的分类器能有效识别有交互关系的蛋白质对,被误分的负类蛋白质对较少。在需要准确识别有交互蛋白质对的系统中,提出的方法能够起到很好的效果。
在签名档集中,正类和负类蛋白质对的签名档的数量相近,一些特征在正类蛋白质对的签名档中较常出现,在负类蛋白质对的签名档中较少出现。这些特征单词有助于识别有交互关系的蛋白质对,因此,采用的dbidf、dsidf和power这三种公式赋予了这些单词较大的权重,使正类蛋白质对的特征向量能明显区别于负类蛋白质对的特征向量,提高蛋白质对交互关系的识别。在正类和负类签名档中分布存在明显差异的单词对于蛋白质对交互关系识别有着很大的作用,给这部分单词赋予一个较大的权值,有助于识别有交互关系的蛋白质对。
4 结束语
区别于其他基于单句的机器学习方法,文中直接以蛋白质对为研究对象,根据现有的PPI数据库构建大规模语料库为依据提取特征,用特征向量表示蛋白质对的关系。根据特征单词在正类和负类蛋白质对签名档中分布的差异,采用不同的权值计算公式研究特征词语的重要性。最后,采用K近邻和SVM分类算法构建分类器,通过文本分类的方法识别目标两个蛋白质对之间是否存在依赖关系。
大量的蛋白质对交互关系信息隐藏在文献中,而基于大规模语料库的PPI识别能充分利用已有的PPI数据,无需进行额外的人工标注,识别结果可直接用于PPI网络的构建。文中着重研究特征词语的重要性,从实验结果可以发现,根据特征单词在不同类别文档中出现的差异,对特征向量根据特征词语重要性进行加权后,PPI的识别结果有了明显提升。
[1]PrasadTSK,GoelR,KandasamyK,etal.Humanproteinreferencedatabase-2009update[J].NucleicAcidsResearch,2009,37:767-772.
[2]KerrienS,Alam-FaruqueY,ArandaB,etal.IntAct-opensourceresourceformolecularinteractiondata[J].NucleicAcidsResearch,2007,35:561-565.
[3]CeolA,AryamontriAC,LicataL,etal.MINT,themolecularinteractiondatabase:2009update[J].NucleicAcidsResearch,2010,38:532-539.
[4]BunescuR,MooneyR,RamaniA,etal.Integratingco-occurrencestatisticswithinformationextractionforrobustretrievalofproteininteractionsfromMedline[C]//Proceedingsoftheworkshoponlinkingnaturallanguageprocessingandbiology:towardsdeeperbiologicalliteratureanalysis.[s.l.]:AssociationforComputationalLinguistics,2006:49-56.
[5]KoikeA,KobayashiY,TakagiT.Kinasepathwaydatabase:anintegratedprotein-kinaseandNLP-basedprotein-interactionresource[J].GenomeResearch,2003,13:1231-1243.
[6] 杨志豪,洪 莉,林鸿飞,等.基于支持向量机的生物医学文献蛋白质关系抽取[J].智能系统学报,2008,3(4):361-369.
[7] 崔宝今,林鸿飞,张 霄.基于半监督学习的蛋白质关系抽取研究[J].山东大学学报:工学版,2009,39(3):16-21.
[8]GrimesGR,WenTQ,MewissenM,etal.PDQWizard:automatedprioritizationandcharacterizationofgeneandproteinlistsusingbiomedicalliterature[J].Bioinformatics,2006,22(16):2055-2057.
[9]AnaniadouS,KellDB,TsujiiJ.Textmininganditspotentialapplicationsinsystemsbiology[J].TrendsinBiotechnology,2006,24(12):571-579.
[10]FundelK,KüffnerR,ZimmerR.RelEx-relationextractionusingdependencyparsetrees[J].Bioinformatics,2007,23(3):365-371.
[11]TemkinJM,GilderMR.Extractionofproteininteractioninformationfromunstructuredtextusingacontext-freegrammar[J].Bioinformatics,2003,19(16):2046-2053.
[12]QianW,FuC,ChengH.Semi-supervisedmethodforextractionofprotein-proteininteractionsusinghybridmodel[C]//Proceedingsofthe2013thirdinternationalconferenceonintelligentsystemdesignandengineeringapplications.[s.l.]:IEEEComputerSociety,2013:1268-1271.
[13] Niu Y,Otasek D,Jurisica I.Evaluation of linguistic features useful in extraction of interactions from PubMed;application to annotating known,high-throughput and predicted interactions in I2D[J].Bioinformatics,2010,26(1):111-119.
[14] Haussler D.Convolution kernels on discrete structures[R].California:University of California at Santa Cruz,1999.
[15] Lodhi H,Saunders C,Shawe-Taylor J,et al.Text classification using string kernels[J].Journal of Machine Learning Research,2002,2(3):419-444.
[16] Bunescu R C,Mooney R J.A shortest path dependency kernel for relation extraction[C]//Proceedings of the conference on human language technology and empirical methods in natural language processing.[s.l.]:Association for Computational Linguistics,2005:724-731.
[17] 封二英,牛 耘,魏 欧,等.基于关系相似性的蛋白质交互自动识别[J].计算机科学,2013,40(6):229-232.
[18] 封二英,牛 耘,魏 欧.基于大规模文本的蛋白质交互关系自动提取[J].计算机应用,2012,32(S1):147-150.
[19] U.S.National Library of Medicine.PubMed[EB/OL].2011.http://www.ncbi.nlm.nih.gov/pubmed/.
[20] Sparck J K.A statistical interpretation of term specificity and its application in retrieval[J].Journal of Documentation,1972,28(1):11-21.
[21] Paltoglou G,Thelwall M.A study of information retrieval weighting schemes for sentiment analysis[C]//Proceedings of the 48th annual meeting of the association for computational linguistics.[s.l.]:Association for Computational Linguistics,2010:1386-1395.
[22] Chang C C,Lin C J.LIBSVM:a library for support vector machines[J].ACM Transactions on Intelligent Systems & Technology,2007,2(3):389-396.
Identification of Protein-protein Interaction Based on Feature Weighted
WU Hong-mei,NIU Yun
(School of Computer Science and Technology,Nanjing University of Aeronautics and Astronautics,Nanjing 210016,China)
In a model characterized by word,if the use of feature word in different categories exists obvious differences,it will have a very important impact on classification.Based on a large-scale corpus,study the effects of different methods of feature weighting on protein-protein interaction identification.Firstly,the signature of a protein pair is obtained by searching large scale biomedical text.Taking the words as the features which describe the relationship between the protein pair,construct Vector Space Model (SVM).Then,select different weighting methods to describe the importance of words.Finally,KnearestneighborandSVMclassifierareappliedtoidentifyPPIs.Accordingtotheexperimentalresults,PPIrecognitionaccuracyandrecallandprecisionhavebeensignificantlyimprovedwhenthefeaturevectorsareweighted.
protein-protein interaction;large-scale corpus;feature weighted;Knearestneighbor;SVM
2015-05-10
2015-08-13
时间:2016-01-26
国家自然科学基金资助项目(61202132,61170043)作者简介:吴红梅(1991-),女,硕士研究生,研究方向为自然语言处理;牛 耘,博士,副教授,CCF会员,研究方向为自然语言处理。
http://www.cnki.net/kcms/detail/61.1450.TP.20160126.1517.026.html
TP
A
1673-629X(2016)02-0114-04
10.3969/j.issn.1673-629X.2016.02.026
