番茄种子RNA 提取及快速检测qRT-PCR模板纯度的方法优化
2016-01-15杨荣超章月琴丁小明
杨荣超, 章月琴, 丰 锋, 丁小明, 齐 飞
(1.广东海洋大学农学院, 广东 湛江524088;2.农业部规划设计研究院设施农业研究所, 北京100026
Real time RT-qPCR (real-time quantitative reverse transcription-PCR)是检测样本中基因表达量的有效方法,具有灵敏、准确、快速、所需样品量小、操作技术简单等诸多优点,是目前检测基因表达的有效方法[1-2]。然而,该方法却受到实验设计、引物、模板的质量、内参的选择及数据分析的方法等多个因素的影响。因此,保证RNA的完整性及纯度在分析基因转录表达丰度的研究中起到重要的作用,然而,有些研究必须利用特定的植物组织,比如植物的种子。但是,植物的种子内含有大量的多糖、贮藏蛋白、脂肪,多数还含有酚、酮等多种次生代谢物质,这些物质给获得完整和高纯度的RNA带来了很大麻烦[3]。
目前,在实际的研究过程中有多种提取种子RNA的方法和试剂盒,但是提取RNA的基本原理是相通的,不同方法之间区别通常只是改变了其中某种试剂,从而有利于某些特殊的成分降解[4-6]。除去通过改变提取试剂来提取不同组织的高质量的RNA外,提取过程中的主要操作同时也起到非常重要的作用。本研究优化了华越洋生物科技有限公司RNA提取试剂盒的主要操作流程,能够从番茄种子中获得大量完整的RNA。另外,排除DNA的干扰和污染也是得到可靠结论的前提和必要条件[7-9]。为了有效排除DNA的污染和干扰,通常利用DNA酶消除DNA,另外,还可以通过设计跨越表达基因内含子的引物,然后再利用PCR扩增的方法来检测模板的纯度。虽然这两种方法应用比较广泛,但仍然具有一定的局限性。针对不同方法的优劣,本研究通过设计跨越内参基因内含子的引物,利用这对引物扩增分析待测样品是否被基因组DNA污染。这种方法为快速从种子中获得高质量的 Real time RT-q PCR模板提供了有效的保证。
1 材料方法
1.1 植物材料
番茄(Solanum lycopersicum)LA 2711(L.esculentumcv.Edkawi LA 2711),由番茄遗传资源中心(Tomato Genetic Research Center,TGRC)提供,中蔬5号(ZS-5),来自中国农业科学院蔬菜花卉研究所。这2种番茄都是栽培品种,果实较大。
1.2 RNA的提取
挑选取2种番茄大小基本一致的种子200粒供试验用,种子直接置于9cm直径培养皿内,培养皿内铺4层滤纸,每天添加3~4mL的灭菌水,保证培养皿内的足够湿度。2种番茄分别播种4皿,每培养皿播种40粒。培养条件为:日温(24±1)℃,夜温(22±1)℃,16h光照/8h黑暗光周期。分别选取吸胀0,12,18,24,36,48h的种子,利用液氮快速保存后储存于-80℃冰箱备用。
种子内富含蛋白质、脂肪及多糖,要获取高质量的RNA比较困难。本研究对华越洋公司RNA提取试剂盒的提取流程进行相应的改进,获得大量并且完整的种子RNA。RNA的提取流程如下:

膜上DNA的消化流程:
1)9μL RNase-free DNase 1+66μL DNase Buffer,在干净的离心管中充分混匀,加入到离心吸附柱的中央,放置20min,且必须放置一段时间,才能够充分去除DNA。12 000g离心1min,弃穿透液。
2)加0.5mL去酶液,轻柔颠倒50次,保证RNA的完整性。12 000g离心1min,弃穿透液。重复上步实验。
3)12 000g离心2min,主要目的是去除残留去酶液,其主要物质是乙醇,否则乙醇会影响RNA的后续应用。
4)50~100μL RNA洗脱液,室温放置5min,12 000g离心1min。
5)再将洗脱液重新加入离心管,室温放置5min,12 000g离心1min,立即使用或-80℃条件存放。
1.3 方法原理
本研究的核心是设计一对跨越内参基因(EF 1α)内含子(78bp)的引物,利用这对引物以不同的模板进行PCR扩增。如果反转录后的cDNA模板被DNA污染,利用这对引物进行PCR扩增,则能够扩增出441bp的片段;如果cDNA模板没有被DNA污染,则扩增出来为363bp的片段。通过这种策略能够快速检测出DNA的污染。
1.4 PCR及qRT-PCR扩增
反转录利用的是TakaRa试剂盒(DRR 037S)。利用跨越内参基因(EF 1α)内含子的引物PCR扩增。PCR的扩增体系20μL,模板分别为:H2O,DNA,RNA转录后的cDNA,没转录的RNA。反应条件:95℃5min,95℃30s,60℃30s,72℃,45s,共30个循环。

表1 各实验所用的引物列表
荧光定量PCR利用96孔罗氏LightCycler 480仪器(Roche Diagnostics,Basel,Switzerland)。20μL的反应体系包括10μL SYBR Primerx Ex Taq(Takara,kyoto,Japan),1.0μL cDNA 模板,上下游引物10 μmol/L各加0.8μL,加ddH2O 6.4μL补足20μL。反应条件:95℃30s;95℃5s,60℃25s,共40个循环。每一个样品3个技术重复,利用番茄PP 2Acs基因的正向引物和反向引物作为内对照,所有候选基因以及PP 2Acs基因引物序列见表1[10],引物由上海生工生物工程有限公司合成。荧光定量数据的计算方法以2-ΔΔCT方式计算[11]。
1.5 数据分析
方差分析采用SAS 8.0软件的ANOVA过程处理,显著性检验采用邓肯法。有关作图采用Excel 2007、SigmaPlot和 Adobe Illustrator CS 5软件。
2 结果分析
2.1 RNA的完整性分析
获得高质量、能正确反映样品中转录丰度的RNA,对于其后续定量结果的准确性起到至关重要的作用。通过改进“华越洋公司”RNA提取试剂盒,变性琼脂糖凝胶电泳结果显示,18S和28S的条带明显而且都比较完整,从图片的亮度可以看出,rRNA比率[28S/18S]≥1(图1)。

图1 RNA的变性琼脂糖凝胶电泳结果分析
2.2 利用跨越内含子的内参基因扩增结果分析

图2 合成cDNA的PCR扩增产物
利用跨越内含子的EF 1α作为引物,分别以灭菌H2O为隐性对照、种子的DNA为阳性对照、RNA和cDNA为模板进行PCR扩增。通过图2(A,B)可看出,以种子基因组DNA为模板扩增出一条441bp的条带;以H2O和RNA为模板没有扩增出条带;以cDNA为模板则扩增出363bp的条带。
2.3 种子萌发过程中2个基因的qRT-PCR表达分析
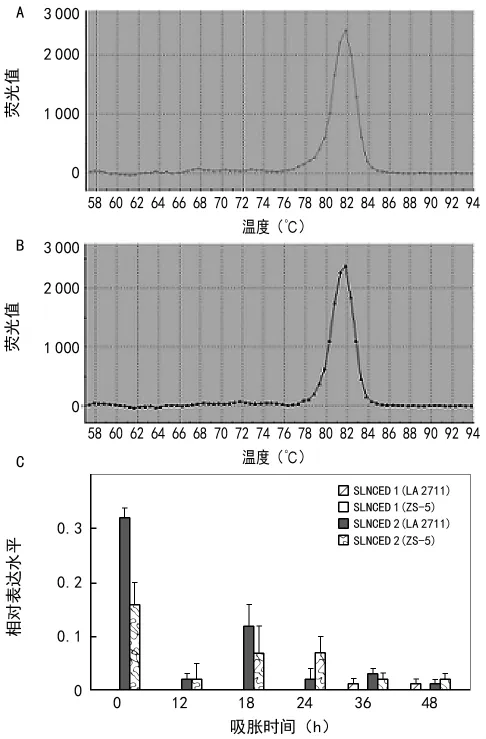
图3 ABA合成基因表达分析
为了验证上述策略的可靠性,对反转录后的样板进行荧光定量PCR分析,通过图3(A,B)在80~90℃温度范围内,2个基因的溶解曲线只有1个单峰,并且出峰的位置在设置的退火温度,说明没有非特异性扩增出来,即 Real time RT-q PCR 的模板没有受到DNA的污染和干扰。在番茄种子萌发的过程中,SlNCED1的相对表达值很低甚至不表达,而SlNCED2的表达较高(图3C)。SlNCED2在2种番茄中的表达规律都是先降低后升高然后再降低的趋势。在0h处SlNCED2的表达量是种子中萌发过程的最大值,该基因在LA 2711的相对表达量是ZS-5的2倍;在12h该基因的表达又降低到最低值,该基因在LA 2711中的相对表达量是ZS-5的0.87倍,为0~12 h,该基因LA 2711中的下降程度显著高于ZS-5。在18h处,LA 2711的表达值又上升,然后又显著降低,但是该基因在18~24h时间内,在ZS-5中的表达一直保持较高的值,而后才降低。
3 讨 论
Real time RT-qPCR 技术是一种快速、简便、准确、灵敏、成本低廉的基因表达分析方法。然而,由于技术的简单性及敏感性使得它在实验过程由于模板的纯度不够而导致结果出现明显的偏离甚至背离。因此,如何控制实验过程中模板的纯度也是研究过程中必须解决的问题。目前,虽然在提取RNA的过程中,为了避免DNA的污染,常常利用DNA的溶解酶溶解RNA样品中的 DNA,但是由于 Real time RT-qPCR非常敏感,即使存在微量的DNA也会对结果造成显著的影响。当然,在DNA污染比较严重的情况下,其溶解曲线也会出现明显的偏离,通过这种途径也可以发现模板被DNA污染,但是这样通常会造成时间及试剂的浪费。在实验过程中,常用的一种方法是每个表达基因的引物跨越内含子,这样利用PCR的方法也能够证明模板的污染情况,但是如果同时分析大量基因时要保证每个基因都跨越内含子的引物,这样保证每对引物的质量困难很大[12-13]。本研究是利用跨越内含子的内参基因引物快速检测已经反转录完成的cDNA是否被DNA污染,这种方法首先可以避免多个表达基因的引物都要跨越内含子给设计引物带来的麻烦。另外,这种方法也是荧光定量分析前快速、有效的方法,这样通常能够避免由于DNA的污染带来的不必要的试剂浪费。当然,目前也有一些学者不推荐利用跨越内含子的引物来检测模板的纯度,主要原因是超过20%人类基因被认为是单一外显子基因[2],因此,利用跨越内含子引物检测也具有一定的局限性。但是模式植物上一些内参基因的结构信息已经非常明确,通过设计看家基因跨越内含子的引物来检测DNA的污染还是非常有效的方法。
[1]Nolan T,Hands R.E,Bustin S.A,et al.Quantification of mRNA using real time PCR[J].Nature Protocols.2006,1(3):1 559-1 582.
[2]Derveaux S,Vandesomple J,Hellemans J.How to do successful gene expression analysis using real time PCR[J].Methods,2010,50(4):227-230.
[3]George N.L,Constantina T,Vassiliki O,et al.Isolation of Tomato Seed Meal,Proteons with Salt solutions[J].Journal of Food Science,1995,60(3):477-482.
[4]Graham G.C.A method for extraction of total RNA from Pinus radiata and other conifers[J].Plant Molecular Biology Reporter,1993,11(1):32-37.
[5]Lewinsohn E,Steele C.L,Croteau R.Simple isolation of functional RNA from woody stems of gymnosperms[J].Plant Molecular Biology Reporter,1994,12(1):20-25.
[6]Tesniere C,Vayda M.E.Method for the isolation of highquality RNA from grape berry tissues without contaminating tannins or carbohydrates[J].Plant Moloecular Biology Reporter,1991,9(3):242-251.
[7]Bernanrd P.S,Wittwer C.T.Real time PCR technology for cancer diagnostics[J].Clinical Chemistry,2002,48(8):1 178-1 185.
[8]Bustin S.A,Mueller R.Real time reverse transcription PCR(RT-qPCR)and its potential use in clinical diagnosis[J].Clinical Science,2005,109(4):365-379.
[9]Bustin S.A,Nolan T.Pitfalls of quantitative real time reverse transcription polymerase chain reaction [J].Journal of Biomolecular Technique,2004,15(3):155-166.
[10]Dekkers B.J,Willems L,Bassel G.W,et al.Identification of reference genes for RT-qPCR expression analysis in Arabidopsis and tomato seeds[J].Plant Cell Physiology,2012,53(1):28-37.
[11]Livak K.J,Schmittgen T.D.Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCTmethod[J].Methods,2001,25(4):402-408.
[12]Fleige S,Pfaffl M.W.RNA integrity and the effect on the real time RT-qPCR performance[J].Molecular Aspects Medicine,2006,27(2-3):126-139.
[13]Huggett J.F,Novak T,Garson J.A,et al.Differential susceptibility of PCR reaction to inhibitors:an important and unrecognised phenomenon[J].BMC Research Notes,2008,28:70.
