β-arrestins与纤维化疾病的研究进展
2016-01-12谷元婧孙妩弋吴晶晶
谷元婧,孙妩弋,张 森,吴晶晶,魏 伟
(安徽医科大学临床药理研究所,抗炎免疫药物教育部重点实验室,安徽 合肥 230032)
β-arrestins与纤维化疾病的研究进展
谷元婧,孙妩弋,张森,吴晶晶,魏伟
(安徽医科大学临床药理研究所,抗炎免疫药物教育部重点实验室,安徽 合肥230032)
中国图书分类号:R-05;R341;R345.57;R364.24;R364.32;R392.11
摘要:β-arrestins是在提纯β-肾上腺素受体激酶(β-adrenergic receptor kinase,β-ARK)的过程中发现的一类重要的接头蛋白和信号调控蛋白,对绝大部分G蛋白偶联受体(G protein-coupled receptor,GPCR)介导的信号转导都具有调节作用。纤维化疾病如肝纤维化、肺纤维化、心血管纤维化等,其发病因素和临床表现各不相同,但最终的病理特征都是细胞外基质(extracellular matrix,ECM)在组织器官内过度沉积。大量研究表明,β-arrestins在纤维化疾病的炎症反应、ECM沉积等作用中发挥重要作用,该文就β-arrestins在纤维化疾病中的研究现状和发展前景作一综述。
关键词:β-arrestins;纤维化疾病;细胞外基质;G蛋白偶联受体;信号转导;受体酪氨酸激酶
β-arrestins是一类重要的细胞内接头蛋白和信号调控蛋白,属于arrestin家族。β-arrestins被认为是G蛋白偶联受体(G protein-coupled receptor, GPCR)的负调控因子,参与受体的脱敏和内化。但随着对其的深入研究发现,β-arrestins不仅可阻断GPCR的信号转导,还影响着酪氨酸激酶受体(tyrosine kinase receptor,TKR)、丝裂原激活的蛋白激酶(mitogen-activated protein kinase,MAPK)等介导的信号转导,在细胞的增殖、凋亡及炎症反应中都发挥了重要作用。纤维化是细胞外基质(extracellular matrix,ECM)过度沉积的病理过程,可能导致各个器官的纤维化疾病形成,若不能得到有效控制,最终可能严重影响人类健康。有关统计资料表明,因各种疾病而致死的病人中,接近45%可以归于组织纤维增生疾病,且纤维化疾病的发病率和致死率呈现不断上升的趋势。纤维化疾病已成为人类生命的一大威胁,研究纤维化疾病的病理机制和有效治疗方法显得日益重要。近年来,研究发现,β-arrestins的异常表达与多种纤维化疾病的发生、发展都有密切联系。因此,β-arrestins在纤维化疾病病理过程中的相关作用以及β-arrestins能否作为纤维化疾病治疗的靶点,都值得深入研究。本文就近年来β-arrestins在纤维化疾病中作用的研究进展作一综述。
1β-arrestins的结构
Arrestin家族是一类分子质量在48~55 ku的蛋白,在哺乳动物体内主要分为两类:一类是广泛表达于各组织的β-arrestins,包括 β-arrestin1(arrestin2)和β-arrestin2(arrestin3);另一类是分布于视网膜,调节光受体的信号转导的arrestin1和arrestin4[1]。此外,还有D-和E-arrestin,具有广泛的组织分布,但还没有具体深入的研究。最初,在研究GPCR的信号转导过程中,首次发现了arrestin在GPCR脱敏中的作用。之后,通过分子克隆的方法,分别从牛脑、大鼠脑互补DNA文库里克隆出了与arrestin类似的蛋白——β-arrestin1、β-arrestin2。
β-arrestins的基本结构由两个反向平行的β-折叠组成,分别是N端结构域和C端结构域,每个β-折叠包含7股肽链,中间由一个含12个氨基酸残基的铰链区连接。两种β-arrestin有78%的氨基酸序列是一致的,区别主要位于C-端。在功能结构域上,β-arrestins的N-端包含Src-SH3结合位点;C-端包含JNK3结合位点,调控GPCR信号通路和MAPK信号通路。C-端还包含网格蛋白(clathrin)、AP2(adaptor protein)结合位点,是β-arrestins参与受体内化的结构基础。通过对β-arrestins的质谱分析发现,有多达337种蛋白质可以与β-arrestins作用,其中能与β-arrestin2作用的蛋白较β-arrestin1多,部分蛋白与两种β-arrestin都可作用[2]。另外,对β-arrestins基因敲除小鼠的研究发现,单独敲除β-arrestin1或β-arrestin2的小鼠可以存活,而同时敲除β-arrestin1和β-arrestin2,小鼠致死,说明β-arrestin1和β-arrestin2在功能上有所重叠[3]。
2β-arrestins与纤维化相关通路
近年来,研究发现β-arrestins不仅可调节GPCR的信号转导,还对许多其它非GPCR信号通路有着重要调节作用(Fig 1)。这些信号通路参与了纤维化疾病的病理进程,β-arrestins通过调节这些信号通路,影响ECM的沉积、炎症反应的激活等,从而对纤维化疾病的发生、发展起作用。
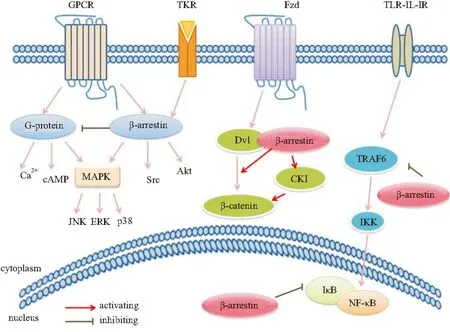
Fig 1 β-arrestins mediated signaling pathways
2.1β-arrestins对GPCR介导的信号转导的调节作用GPCR是七次跨膜受体,是一类重要的细胞表面受体超家族,能够转导激素、神经递质、趋化因子以及光线等物理、化学的细胞外信号。β-arrestins作为GPCR信号的负调控分子,在GPCR信号转导的调控中发挥重要作用。GPCR信号途径包括G蛋白途径和β-arrestins途径[4]。GPCR可能存在几种活性构象,不同的配体引发受体构象的改变不同,有些构象利于 G蛋白结合,有些构象则利于β-arrestins结合,从而激活特异的下游信号。G蛋白信号途径为G蛋白-第二信使依赖性级联反应,激动剂结合到GPCR上,激活G蛋白信号途径,包括钙离子等第二信使系统、腺苷酸环化酶 (adenylcyclase, AC)、MAPK等。β-arrestins在GPCR激动剂刺激下,与磷酸化的GPCR结合后,改变受体构象,促进受体与G蛋白解偶联,即受体脱敏。活化的受体被定向降解或再循环到细胞表面,从而导致受体信号转导的减弱或终止。β-arrestins偏爱性配体则主要激活β-arrestins途径,以G蛋白非依赖的信号通路方式激活信号转导分子,如MAPK、Src蛋白酪氨酸激酶和蛋白激酶B(protein kinase B,PKB/Akt)等。已发现的β-arrestins偏爱性配体包括血管紧张素Ⅱ、肾上腺素、内皮素、趋化因子等。
2.2β-arrestins对非GPCR介导的信号转导的调节作用TKR是一类重要的信号蛋白,也是一类受β-arrestins调节的受体蛋白。TKR包括表皮生长因子受体(epidermal growth factor receptor,EGFR)、胰岛素样生长因子受体1(insulin-like growth factor 1 receptor,IGF-1R)等。TKR的主要信号转导是Ras/Raf/MAPK途径和磷脂酰肌醇-3激酶 (phosphatidylinositol-3 kinase,PI3K)/Akt途径。胰岛素样生长因子1(insulin-like growth factor 1,IGF-1)可刺激胶原蛋白合成、肌样成纤维细胞增殖,大量证据显示肠道过度表达IGF-l会导致肠纤维化的发生,而β-arrestin1可影响IGF-lR内化/泛素化。β-arrestin1结合并介导了IGF-lR内吞,可促进ERK活化,正性调节MAPK。此外,β-arrestin1与IGF-lR结合后,能激活PI3K,进而活化Akt,对PI3K途径有正性调控作用,表现出抗凋亡的作用[5]。表皮生长因子(epidermal growth factor,EGF)是一种强烈的促多种细胞增生的有丝分裂原。在心脏中,依赖β-arrestins的EGFR转位激活可以抑制β-AR持续激动引起的心肌细胞凋亡和病理性心脏重塑,因而对心脏有保护作用[6]。
2.3β-arrestins调节Wnt/β-连环蛋白信号途径Wnt与其受体卷曲蛋白(Frizzled)结合,胞质中的散乱蛋白(dishevelled,Dvl)被募集至胞膜附近,Dvl的激活抑制了糖原合成激酶 3β、轴蛋白等形成的复合产物,减少复合产物对于β-连环蛋白(β-catenin)的降解,从而激活Wnt/β-catenin通路。而β-arrestins能够与Dvl结合,通过破坏复合产物的形成抑制β-catenin降解;β-arrestins还能够与轴蛋白结合,与Dvl一起形成三聚体复合物,参与信号传递[7]。过多的β-catenin积累并转移到细胞核内,与T细胞因子相结合,在其他因子的辅助下协同激活靶基因的转录,诱导疾病的发生。Henderson等[8]研究发现,使用Wnt/β-catenin抑制剂ICG-001能够明显抑制β-catenin信号通路,缓解由博来霉素诱导的小鼠肺纤维化。敲除β-arrestins的肺纤维化小鼠模型具有较高存活率[9]。此外,缺少β-arrestins还可以使小鼠胚胎成纤维细胞Dvl活化减少,β-catenin的信号传递受抑制[7]。推测β-arrestins可能通过激活Wnt通路参与纤维化疾病进程。
2.4β-arrestins对MAPK信号通路的调节作用MAPK是一组能被不同的细胞外刺激信号,如细胞因子、神经递质、激素、细胞应激及细胞黏附等激活的丝氨酸-苏氨酸蛋白激酶,可磷酸化各种各样的蛋白底物,调节细胞增殖、分化和凋亡。β-arrestins在调节MAPK级联反应方面的作用是研究最为广泛的信号转导机制。细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)可通过G蛋白和β-arrestins两种途径激活,且作用强度不同。G蛋白依赖的ERK的活化看起来是迅速的、瞬时的;β-arrestins依赖的ERK的激活是缓慢的,更加持久。原因可能是两者激活ERK从细胞核排出、并且被限制在细胞质中的能力不同。胞质中的ERK的激活和磷酸化,使许多胞质中的ERK底物,以及一些转录因子转位至细胞核,调控细胞生长、增殖、分化,以及Ⅰ、Ⅲ型胶原的合成等效应,参与多种纤维化疾病。因此,β-arrestins作为支架蛋白,通过细胞核、细胞质中转录因子的磷酸化和激活影响了疾病反应[10]。在多种纤维化疾病如特发性肺纤维化、肝纤维化患者中均观察到JNK通路的异常激活。β-arrestins在JNK3中也起支架蛋白的作用,使JNK3停留在细胞质中时间延长,从而延长JNK3活化状态的寿命,使Raf、MEK和ERK联系起来。
2.5β-arrestins对NF-κB信号通路的调节作用核转录因子κB(nuclear factor-κB,NF-κB)是普遍存在于细胞质中的一种快反应转录因子,参与炎症反应相关的纤维化疾病。NF-κB和NF-κB的抑制性蛋白IκB结合而呈非活性状态,β-arrestin2可直接阻止IκB的磷酸化和降解,从而减弱了NF-κB活化和NF-κB 目的基因的转录,由此负向调节NF-κB信号通路[11]。此外,在Toll样受体-白细胞介素1受体(TLR-IL-1R)介导的TLR-NF-κB通路中,肿瘤坏死因子受体活化因子6(tumor necrosis factor receptor-associated factor 6,TRAF6)是TLR激活下游信号共用的信号转导分子。β-arrestins与TRAF6相互作用后,β-arrestins抑制TRAF6寡聚化,从而使免疫应答关键信号系统TLR-IL-1R不能引发信号转导,抑制NF-κB和AP -1介导的转录及其他炎症相关因子[12]。
2.6 β-arrestins对PI3K/Akt信号通路的调节作用Akt是保护细胞存活、抑制细胞凋亡的重要效应器。对肝纤维化中的肝星状细胞(hepatic stellate cell,HSC)研究发现,Akt的磷酸化水平和I型胶原的mRNA、蛋白表达水平均升高,而阻断PI3K活性能够减少I型胶原的表达;在特发性肺纤维化病人中PI3K/Akt通路异常活化,进而促进成纤维细胞增殖[13]。而在研究小鼠胚胎成纤维细胞时发现,IGF-1R通过β-arrestin1激活PI3K/Akt通路,促进细胞增殖[5]。
3β-arrestins在纤维化疾病进程中的作用
纤维化是组织和分子内各种糖蛋白、胶原等组成的ECM过度沉积形成的,最终可能导致永久型损伤甚至死亡,可发生于多种器官,如肝脏、肾脏、胆囊等。这种病理过程也是许多慢性自身免疫病的特征,如硬皮病、风湿性关节炎、溃疡性结肠癌、系统红斑狼疮等。纤维化本质在于保持ECM的体内平衡,所以,在早期是一个可逆的过程。目前,对于治疗纤维化疾病仍然没有非常有效的方法,只能通过抑制ECM生成、加快ECM降解以及抵抗炎症作用来预防和抑制纤维化生成。近年来,越来越多的研究发现,β-arrestins与纤维化形成有着密切联系,影响着纤维化进程。
3.1肝纤维化有研究表明,在猪血清诱导的肝纤维化大鼠模型中,随着造模时间延长,纤维化肝组织中β-arrestin2的表达较正常大鼠肝组织明显增多,而β-arrestin1的表达无明显变化。活化的HSC是肝纤维化ECM的主要来源细胞,平滑肌肌动蛋白(α-smooth muscle actin,α-SMA)是活化HSC的表面标志。免疫荧光检测发现,肝纤维化大鼠模型进展过程中,肝组织β-arrestin2与α-SMA的表达呈正相关,且随造模时间延长表达增加,提示β-arrestin2在肝纤维化发展过程中起重要作用。用血小板衍生生长因子-BB(platelet-derived growth factor-BB,PDGF-BB)刺激HSC细胞,β-arrestin2表达升高。HSC转染siRNA-β-arrestin2后,HSC的增殖明显受到抑制,并且ERK1/2通路的激活也受到抑制[14];同时,细胞内抑制细胞凋亡的Bcl-2表达下调,促进细胞凋亡的Bax表达上调,HSC凋亡率明显升高。提示β-arrestin2的缺失可以抑制ERK1/2通路的激活和Bcl-2/Bax比率,抑制HSC的增殖,促进其凋亡[15]。
另外,对原发性胆汁肝硬化患者的研究发现,与正常人群相比,患者的外周血单核细胞中NF-κB和AP-1的活性明显降低,而β-arrestin1 mRNA和蛋白水平明显升高,这可能与β-arrestin1抑制AP-1 和 NF-κB的激活有关[16]。慢性乙型肝炎(hepatitis B virus,HBV)、丙型肝炎(hepatitis C virus,HCV)是导致肝硬化的主要原因。实验研究结果表明,在HBV、HCV患者的活组织切片中,编码β-arrestin2的基因ARRB2在肝炎患者中表达较高[17]。提示β-arrestins对肝硬化的发生发展也有影响。
3.2肺纤维化研究者用野生型小鼠、β-arrestin1敲除的小鼠和β-arrestin2敲除的小鼠建立博来霉素诱导的肺纤维化模型,结果发现,野生型小鼠的死亡率在21 d内达到50%,而β-arrestin1、β-arrestin2敲除的小鼠都具有较高的存活率。组织学检查发现野生型小鼠胶原大量沉积,而β-arrestin1、β-arrestin2敲除的小鼠沉积较少。同时,β-arrestin1、β-arrestin2敲除的小鼠的原始肺纤维母细胞表现出较弱的迁徙能力[9]。提示下调β-arrestins可能是治疗肺纤维化的新靶点。
3.3肾脏纤维化各种原因引起的慢性肾脏疾病最终进展成为终末期肾衰竭,其实质都是肾脏纤维化和硬化,导致有效肾单位丧失和肾功能的下降。糖尿病肾病是导致肾脏末期疾病的重要因素, nephrin蛋白的缺失和蛋白尿的发生则是肾衰竭的决定性因素。免疫共沉淀实验表明,在HEK293T细胞中,β-arrestin2与nephrin蛋白相互作用,并且促进nephrin蛋白的内吞,β-arrestin2越多,细胞表面的nephrin蛋白越少[18],最终终止信号转导。在高糖环境下,过表达β-arrestin2的HEK293T细胞加蛋白激酶Cα(protein kinase Cα,PKCα)抑制剂,发现β-arrestin2和nephrin蛋白的相互作用下调。提示在此环境中两者的相互作用受到PKCα的调节[19],提示下调β-arrestin2可能有利于抑制疾病的发展。
3.4肠壁纤维化肠道慢性炎症会导致修复性损伤和纤维化,肠纤维化的发展与炎症发展息息相关,是炎症肠病反复发作的临床问题。炎症性肠病是受多因子调节的胃肠道炎症,以溃疡性结肠炎(ulcerative colitis,UC)为代表。肠道在反复的慢性炎症和过度损伤修复下,可引起肠壁的纤维化。
NF-κB是肠道慢性炎症发病的一个重要因素,可上调肠黏膜中致炎细胞因子表达,加重肠黏膜的炎症。在小鼠UC模型中,β-arrestin1敲除小鼠的结肠长度长于野生型小鼠,检测小鼠血浆和结肠组织发现,β-arrestin1缺失小鼠中IL-6的表达明显低于野生型小鼠,IL-10、IL-22水平明显升高[20]。在UC模型的结肠黏膜组织和脾脏淋巴细胞中NF-κB p65表达水平明显上升,而β-arrestin2表达明显下降,其机制与β-arrestin2与IκB作用减弱,激活NF-κB信号转导通路,从而参与UC的形成[21]。提示β-arrestin1和β-arrestin2在溃疡性结肠炎中发挥相反的作用。
3.5心血管纤维化血管硬化主要指动脉硬化,动脉粥样硬化病变的形成是动脉对内膜损伤作出的炎症-纤维增生性反应的结果。而平滑肌细胞(smooth muscle cell, SMC)的增殖与迁移是动脉粥样硬化形成关键。有研究者使用单独敲除低密度脂蛋白受体(low density lipoprotein receptor,LDLR)的小鼠、及同时敲除LDLR和β-arrestin2的小鼠,研究发现与单独敲除LDLR组比较,同时敲除LDLR和β-arrestin2组小鼠动脉粥样硬化发病率明显降低,且动脉SMC增殖明显减少。实验分别观察了正常、β-arrestin1敲除、β-arrestin2敲除3组小鼠颈动脉内皮剥脱后剥脱处内膜增生情况,β-arrestin1敲除组升高,β-arrestin2敲除组降低。颈动脉受损后,SMC的ERK信号通路在β-arrestin1敲除组升高,β-arrestin2敲除组降低。提示,β-arrestins2通过激活ERK通路促进SMC的增殖与迁移,形成粥样硬化,而β-arrestins1发挥相反作用[22]。
在研究心肌纤维化时发现,长期使用β-AR阻断剂可诱导野生型小鼠心脏产生血管周的纤维化,定量mRNA分析发现心肌细胞中纤维化基因的表达也随之增加。但β-arrestin2敲除的小鼠使用β-AR阻断剂3个月后,没有在心脏血管周发生纤维化,而与之有关的纤维化基因表达也被抑制[23]。提示下调β-arrestin2可能抑制心肌纤维化。
3.6骨髓纤维化慢性粒细胞性白血病(chronic myelogenous leukemia,CML)是骨髓及骨髓外增殖肿瘤的一种,基本特点是染色体结构不稳定,导致BCR与ABL原癌基因融合成为BCR-ABL致癌基因。在CML的自然病程中,会普遍出现不同程度的骨髓纤维化。Fereshteh等[24]在β-arrestin1敲除、β-arrestin2敲除和野生型小鼠中分别转入BCR-ABL基因,发现敲除β-arrestin2小鼠存活率较高。通过转染针对β-arrestin2的shRNA进入CML干细胞,β-arrestin2的低表达降低了CML干细胞的克隆增殖。下调β-arrestin2有效降低了CML细胞Wnt/β-连环蛋白通路的激活,提示β-arrestin2可能通过激活Wnt/β-连环蛋白通路参与CML。
3.7中枢神经系统纤维化多发性硬化(multiple sclerosis,MS)是中枢神经系统髓鞘性疾病中最常见的一种类型,主要发生在中枢神经白质,是脱髓鞘所致胶原纤维化。MS一般被认为是由髓磷脂蛋白激活的T细胞所调节。对MS患者外周血研究发现,T细胞在MS中的增殖反应可被包括β-arrestin1在内的3种非髓磷脂蛋白激活[25]。同时外周血中编码β-arrestin1的基因ARRB1在CD4+T细胞中表达高于正常组[26]。此外,MS患者脑组织β-arrestin1表达较正常人脑组织明显升高。G蛋白偶联的腺苷A1受体(A1 adenosine receptor,A1AR)有着抗炎、保护组织的作用,在人单核细胞中过表达β-arrestin1,降低A1AR表达。糖皮质激素作用人单核细胞后,A1AR上调,β-arrestin1表达下调[27]。提示β-arrestin1可能通过促进炎症相关反应参与了MS。
3.8胆囊纤维化胆囊纤维化(cystic fibrosis,CF)是由于囊纤维化跨膜调节因子(cystic fibrosis transmembrane regulator,CFTR)缺失引起的炎症反应造成的,最近研究发现与胆固醇的沉积也有关联。有研究者建立CFTR缺失小鼠模型,模型组中CF细胞的β-arrestin2高表达。油脂染色实验表明,在过表达β-arrestin2的CF细胞核周围有明显胆固醇沉积[28]。对CF细胞的研究发现,cAMP反应元件(cAMP-response element binding protein,CREB)可激活cAMP,上调cAMP通路可导致胆固醇沉积,CF细胞中β-arrestin2的表达与CREB的表达呈正相关[29],从而促进胆固醇的沉积。
4结语
纤维化疾病可发生于机体多种重要器官,如肝脏、肺、肾脏、心脏等,对机体造成了重大危害,严重者可危及生命,而炎症反应和ECM的沉积对纤维化的形成起重要作用。在肝纤维化、心肌纤维化、肺纤维化等纤维化疾病中,β-arrestins mRNA的水平和β-arrestins的异常都说明β-arrestins是各器官纤维化发生、发展的重要环节。β-arrestins通过调节GPCR、细胞因子、激酶等的表达,参与调节炎症和ECM沉积等多条与纤维化相关的信号通路,在器官纤维化过程中发挥着不可忽视的作用。根据其特点对其进行更具体化的研究或许可以拓展研究和治疗纤维化及纤维化相关疾病的思路。由于纤维化细胞因子机制研究中的复杂性,特别是细胞因子网络中各细胞因子间的相互作用,各通路间的相互激活,及其对纤维化发生进展过程的影响,需进一步深入研究β-arrestins在纤维化相关疾病的作用机制。
参考文献:
[1]Reiter E, Lefkowitz R J. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling[J].TrendsEndocrinolMet, 2006,17(4):159-65.
[2]Xiao K, McClatchy D B, Shukla A K, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis[J].ProcNatlAcadSciUSA, 2007,104(29):12011-6.
[3]DeWire S M, Ahn S, Lefkowitz R J, et al. Beta-arrestins and cell signaling[J].AnnuRevPhysiol, 2007,69:483-510.
[4]Violin J D, Lefkowitz R J. Beta-arrestin-biased ligands at seven-transmembrane receptors[J].TrendsPharmacolSci, 2007,28(8):416-22.
[5]Povsic T J, Kohout T A, Lefkowitz R J. Beta-arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis[J].JBiolChem, 2003,278(51):51334-9.
[6]Noma T, Lemaire A, Naga P S, et al. Beta-arrestin-mediated betal-adrenergic receptor transactivation of the EGFR confers cardioprotection[J].JClinInvest, 2007,117(9):2445-58.
[7]Bryja V, Gradl D, Schambony A, et al. Beta-arrestin is a necessary component of Wnt/beta-catenin signalinginvitroandinvivo[J].ProcNatlAcadSciUSA, 2007,104(16):6690-5.
[8]Henderson W J, Chi E Y, Ye X, et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis[J].ProcNatlAcadSciUSA, 2010,107(32):14309-14.
[9]Lovgren A K, Kovacs J J, Xie T, et al. beta-arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix[J].SciTranslMed, 2011,3(74):23r-74r.
[10]Houslay M D, Baillie G S. Beta-arrestin-recruited phosphodiesterase-4 desensitizes the AKAP79/PKA-mediated switching of beta2-adrenoceptor signalling to activation of ERK[J].BiochemSocTrans, 2005,33(Pt 6):1333-6.
[11]Luan B, Zhang Z, Wu Y, et al. Beta-arrestin2 functions as a phosphorylation-regulated suppressor of UV-induced NF-kappaB activation[J].EMBOJ, 2005,24(24):4237-46.
[12]Wang Y, Tang Y, Teng L, et al. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling[J].NatImmunol, 2006,7(2):139-47.
[13]Xia H, Diebold D, Nho R, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis[J].JExpMed, 2008, 205(7): 1659-72.
[14]Sun W Y, Song Y, Hu S S, et al. Depletion of beta-arrestin2 in hepatic stellate cells reduces cell proliferation via ERK pathway[J].JCellBiochem, 2013,114(5):1153-62.
[15]宋杨, 孙妩弋, 胡姗姗, 等. siRNA沉默β-arrestin2促进肝星状细胞凋亡[J]. 中国药理学通报, 2012,28(5):612-6.
[15]Song Y, Sun W Y, Hu S S, et al. Small interfering RNA targeting β-arrestin2 promoted apoptosis of hepatic stellate cells[J].ChinPharmacolBull,2012,28(5):612-6.
[16]Hu Z, Huang Y, Liu Y, et al. beta-Arrestin 1 modulates functions of autoimmune T cells from primary biliary cirrhosis patients[J].JClinImmunol, 2011,31(3):346-55.
[17]Ciesla A, Kusmider M, Faron-Gorecka A, et al. Intrahepatic expression of genes related to metabotropic receptors in chronic hepatitis[J].WorldJGastroenterol, 2012,18(31):4156-61.
[18]Quack I, Rump L C, Gerke P, et al. beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity[J].ProcNatlAcadSciUSA, 2006,103(38):14110-5.
[19]Quack I, Woznowski M, Potthoff S A, et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia[J].JBiolChem, 2011,286(15):12959-70.
[20]Lee T, Lee E, Irwin R, et al. beta-Arrestin-1 deficiency protects mice from experimental colitis[J].AmJPathol, 2013,182(4):1114-23.
[21]Fan H, Liao Y, Tang Q, et al. Role of beta2-adrenoceptor-beta-arrestin2-nuclear factor-kappaB signal transduction pathway and intervention effects of oxymatrine in ulcerative colitis[J].ChinJIntegrMed, 2012,18(7):514-21.
[22]Kim J, Zhang L, Peppel K, et al. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration[J].CircRes, 2008,103(1):70-9.
[23]Nakaya M, Chikura S, Watari K, et al. Induction of cardiac fibrosis by beta-blocker in G protein-independent and G protein-coupled receptor kinase 5/beta-arrestin2-dependent signaling pathways[J].JBiolChem, 2012,287(42):35669-77.
[24]Fereshteh M, Ito T, Kovacs J J, et al. beta-Arrestin2 mediates the initiation and progression of myeloid leukemia[J].ProcNatlAcadSciUSA, 2012,109(31):12532-7.
[25]Forooghian F, Cheung R K, Smith W C, et al. Enolase and arrestin are novel nonmyelin autoantigens in multiple sclerosis[J].JClinImmunol, 2007,27(4):388-96.
[26]Shi Y, Feng Y, Kang J, et al. Critical regulation of CD4+T cell survival and autoimmunity by beta-arrestin 1[J].NatImmunol, 2007,8(8):817-24.
[27]Tsutsui S, Vergote D, Shariat N, et al. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells[J].FASEBJ, 2008,22(3):786-96.
[28]Manson M E, Corey D A, Bederman I, et al. Regulatory role of beta-arrestin-2 in cholesterol processing in cystic fibrosis epithelial cells[J].JLipidRes, 2012,53(7):1268-76.
[29]Manson M E, Corey D A, White N M, et al. cAMP-mediated regulation of cholesterol accumulation in cystic fibrosis and Niemann-Pick type C cells[J].AmJPhysiolLungCellMolPhysiol, 2008,295(5):L809-19.
网络出版时间:2015-1-9 13:37网络出版地址:http://www.cnki.net/kcms/doi/10.3969/j.issn.1001-1978.2015.02.006.html
Research progress of β-arrestins in fibrotic diseases
GU Yuan-jing, SUN Wu-yi, ZHANG Sen, WU Jing-jing,WEI Wei
(InstituteofClinicalPharmacology,AnhuiMedicalUniversity,KeyLaboratoryofAntiinflammatory
andImmuneMedicine,MinistryofEducation,Hefei230032,China)
Abstract:β-arrestins, a kind of important adaptor protein and signal transduction protein found in the purification process of β-adrenergic receptor kinase (β-ARK),were first identified as proteins that have the ability to desensitize G protein-coupled receptors (GPCR). Fibrosis is defined by the overgrowth, hardening,and scarring of various tissues and is attributed to excess deposition of extracellular matrix(ECM) components including colla-gen. A large number of studies have shown that β-arrestins play an important role in the process of fibrotic diseases,involved in inflammatory response and excess deposition of ECM. This review discusses the research status and development prospects of β-arrestins-mediated fibrotic diseases.
Key words:β-arrestins;firotic disease;extracellular matrix;G protein coupled receptor;signal transduction;receptor tyrosine kinase
通讯作者孙妩弋(1981-),女,博士,副教授,硕士生导师,研究方向:肝脏药理学,,Tel:0551-65161206,E-mail:sunwuyi51@hotmail.com; 魏伟(1960-),男,博士,教授,博士生导师,研究方向:临床药理学,,Tel:0551-65161209,E-mail:wwei@ahmu.edu.cn
作者简介:谷元婧(1991-),女,硕士生,研究方向:肝脏药理学,E-mail:guyuanjing59@hotmail.com;
基金项目:国家自然科学基金资助项目(No 81300332,81173075,81330081);高等学校博士学科点专项科研基金资助项目(No 2011342012002,20123420110003);安徽省高等学校省级自然科学研究项目(No KJ2012A153,KJ2014A119);安徽省自然科学基金资助项目(No 1308085QH130)
收稿日期:2014-10-17,修回日期:2014-11-20
文献标志码:A
文章编号:1001-1978(2015)02-0170-06
doi:10.3969/j.issn.1001-1978.2015.02.006
