Bacillaene酮还原酶结构域的异源表达及底物特异性分析
2015-12-29孙潇慧车程川季俊杰郑舰艇杨革
孙潇慧,车程川,季俊杰,郑舰艇,杨革
Bacillaene酮还原酶结构域的异源表达及底物特异性分析
孙潇慧1,2*,车程川1*,季俊杰2,郑舰艇2,杨革1
1 曲阜师范大学 生命科学学院,山东 曲阜 273165 2 中国科学院过程工程研究所 生化工程国家重点实验室,北京 100190

孙潇慧, 车程川, 季俊杰, 等. Bacillaene酮还原酶结构域的异源表达及底物特异性分析. 生物工程学报, 2015, 31(9): 1355–1362.Sun XH, Che CC, Ji JJ, et al. Heterologous expression and substrate specificity of ketoreductase domain in Bacillaene polyketide synthase. Chin J Biotech, 2015, 31(9): 1355–1362.
Bacillaene生物合成过程中,聚酮合酶第一个延伸模块的酮还原酶结构域 (BacKR1) 既催化α酮基的还原,也催化β酮基的还原,具有天然的底物宽泛性。为进一步研究该结构域的底物特异性,在大肠杆菌中对其进行了异源表达。体外酶学分析表明BacKR1可以催化聚酮类底物 (±)-2-甲基-3-氧代戊酸-乙酰半胱胺硫酯外消旋体的立体选择性还原,仅生成4种非对映异构体中的一种,此外BacKR1还可以催化环己酮和对氯苯乙酮等非聚酮类底物的还原,暗示了聚酮合酶中酮还原酶结构域作为生物催化剂的潜力。
聚酮,酮还原酶,异源表达,立体选择性,底物特异性
聚酮类化合物是由微生物、植物等产生的一大类具有活性和结构多样性的天然产物。许多聚酮类化合物已经被开发为抗癌、抗感染、抗病毒和抗寄生虫等药物,用于人类疾病的治疗[1-2]。聚酮类天然产物的生物合成过程类似于脂肪酸的合成,由短链羧酸通过连续的脱羧聚合而形成。然而与脂肪酸合成机制一个明显的区别是,在聚酮合酶 (Polyketide synthase,PKS) 模块中常常有一个修饰结构域酮还原酶 (Ketoreductase,KR),从而在聚酮终产物中留下许多β-羟基手性中心。此外聚酮合成前体常常是丙酸,导致聚酮内酯环上具有多个α-甲基手性中心[3]。聚酮的生物合成机制研究表明,KR结构域同时控制产物中α碳和β碳的立体构型,相应地KR结构域被分为A1、A2、B1、B2四种不同类型 (图1A)[4]。KR结构域可以在大肠杆菌中异源表达并且保留催化聚酮类底物还原的能力[5]。此外人们还发现,某些KR结构域,如红霉素聚酮合酶第一个延伸模块中的EryKR1,可以催化非聚酮底物如环己酮、二辛酮的还原,表明KR结构域具有作为生物催化剂的潜力[6-7]。
Bacillaene是枯草芽胞杆菌中由PKS和非核糖体肽合成酶 (Nonribosomal peptide-synthetase,NRPS) 共同催化合成的一种抗生素[8-9]。其PKS第一个延伸模块中的KR结构域 (BacKR1) 既可以催化β酮基的还原,也可以催化α酮基的还原 (图1B)[10]。为研究其底物特异性,利用大肠杆菌进行异源表达。体外分析显示BacKR1可以催化外消旋聚酮类底物 (±)-2-甲基-3-氧代戊酸-乙酰半胱胺硫酯 ((±) 2-methyl-3-oxopentanoate N-acetyl cysteamine thioester,MOP-NAC) 的立体选择性还原,只生成4种非对映异构体中的一种,表明BacKR1可以作为生物催化剂合成手性纯的α-甲基-β-羟基酯作为聚酮合成的前体。对8种非聚酮底物的还原表明BacKR1可以催化环己酮和对氯苯乙酮的还原,且与EryKR1相比具有不同的底物谱,暗示了聚酮合酶KR结构域作为生物催化剂的潜力。

图1 酮还原酶催化的酮基还原反应[4,10]
1 材料与方法
1.1 材料
质粒pET28a、菌株BL21 (DE3)、JM109和枯草芽胞杆菌为本实验室保存。LB培养基(成分 (1 L):胰蛋白胨10 g,酵母提取物5 g,NaCl 10 g) 用于培养;BPY培养基(成分 (1 L):牛肉提取物5 g,蛋白胨10 g,酵母提取物5 g,葡萄糖5 g,NaCl 5 g) 用于培养枯草芽胞杆菌。抗生素及诱导剂使用浓度:50 µg/mL硫酸卡那霉素 (Kana),50 µg/mL氯霉素 (Cl),0.1 mmol/L异丙基-b-D-硫代半乳糖苷 (IPTG)。
KOD Plus DNA聚合酶购自TOYOBO公司。T4 DNA连接酶和DNA限制性内切酶购自NEB。Ni-NTA-Sepharose填料购自北京梦怡美生物科技有限公司。化学试剂NADP+和NADPH均购自Sigma-Al-drich公司。环己酮、苯乙酮、苯丙酮、对氯苯乙酮、4-氟苯乙酮、2-辛酮、苯丙酮酸、乙酰乙酸乙酯购自北京中科助研商贸有限公司。二聚酮底物类似物 (±)-2-甲基-3-氧代戊酸-乙酰半胱胺硫酯由本实验室参照文献报导合成[11]。PCR扩增引物合成和DNA测序由生工生物工程 (上海) 股份有限公司完成。
1.2 方法
1.2.1 BacKR1异源表达载体的构建
为了便于蛋白纯化,构建表达载体pET28a-BacKR1。以枯草芽胞杆菌的基因组为模板,利用引物5′-ATCGTAATCGAACGC TTAATGCTTGAACCGGTGT-3′和5′-TGATTCG ATATCCTTGATCCTGATCCGCCTTTCTC-3′ (下划线表示Ⅰ和RⅠ酶切位点) PCR得到含有完整BacKR1的基因片段。用Ⅰ和RⅠ酶切BacKR1片段和pET28a,将酶切得到的载体和目的片段连接。连接产物转化JM109,对得到的克隆进行DNA序列测序,筛选得到构建正确的pET28a-BacKR1。
1.2.2 BacKR1的蛋白表达与纯化
将pET28a-BacKR1表达质粒转化至BL21 (DE3)。挑取单克隆,在含50 µg/mL硫酸卡那霉素的LB 培养基中37 ℃、220 r/min培养至600为0.4–0.6,加入IPTG至0.1 mmol/L,16 ℃、220 r/min诱导表达16 h。离心收集菌体重并悬于50 mmol/L Tris、0.5 mol/L NaCl缓冲液(pH 7.5),超声破碎后离心取上清。亲和层析得到的蛋白进一步用分子筛 (Superdex 200,GE) 进行纯化,得到纯度>95%的蛋白。
1.2.3 外消旋MOP-NAC的合成与还原
化学合成:半胱胺盐酸盐 (1.0 mol),氢氧化钾 (1.5 mol) 和碳酸氢钠 (3.0 mol) 溶于水中常温下搅拌,然后分批滴加乙酸酐 (0.9 mol),滴加时间超过5 min,常温下反应15 min处理得到无色透明油状液体 (化合物1)。三乙胺 (0.8 mL) 溶于10 mL的二氯甲烷,然后分批加入丙酰氯 (0.5 mL),加入时间超过15 min,常温下反应1 h处理得到黄色油状液体 (化合物2)。化合物2 (2.1 mol) 溶于二氯甲烷中,然后加入化合物1 (1.0 mol) 和几滴三乙胺,混合物在常温下反应30 min处理得到外消旋的MOP-NAC (化合物3) (图2)。
化学还原:MOP-NAC (1.0 mol) 溶于四氢呋喃中,加入硼氢化钠 (2.0 mol) 后0 ℃下反应4 h得到无色油状液体 (化合物4) (图2)。化合物4溶于80%正己烷和20%乙醇,根据文献报道,利用OC-H手性柱 (250 mm×4.6 mm,大赛璐药物手性技术有限公司) 进行分离,产物洗脱顺序如图4所示,检测波长为235 nm,流动相为93%的正己烷和7%的乙醇,流速0.8 mL/min[11]。
1.2.4 外消旋MOP-NAC的酶法还原
将纯化得到的酶加入到反应体系中 (总体积为0.2 mL,含有20 µmol/L BacKR1,100mmol/LNADP+,50 mmol/L D-葡萄糖,5 µmol/L葡萄糖脱氢酶 (Glucose dehydrogenase,GDH),50 mmol/L HEPES,10%甘油,100 mmol/L NaCl和1 mmol/L MOP-NAC),30 ℃反应过夜。
为确定立体构型,反应产物经乙酸乙酯萃取,真空抽干,残留物用80%正己烷和20%乙醇溶解。采用上述条件,利用OC-H手性柱分离产物,并通过与化学还原的4种产物进行比较,确定产物构型[11]。

图2 (±)-2-甲基-3-氧代戊酸-乙酰半胱胺硫酯的合成路线及化学还原[11]
1.2.5 非天然底物的还原
利用NADPH在340 nm的吸收检测BacKR1还原非天然底物的能力。反应体系为25 µL,含20 µmol/L BacKR1,1 mmol/L NADPH,50 mmol/L HEPES,10%甘油,100 mmol/L NaCl和1 mmol/L底物,30 ℃反应15 min,测定340的下降,以不加酶的反应液作为对照。选用底物为环己酮、苯乙酮、苯丙酮、对氯苯乙酮、4-氟苯乙酮、2-辛酮、苯丙酮酸、乙酰乙酸乙酯。
2 结果与分析
2.1 表达载体构建及蛋白表达
PKS是分子量巨大 (常大于600 kDa) 的多结构域、多模块酶复合物,表达纯化和酶学分析困难[12]。近十年来的结构生物学研究和体外功能分析表明,PKS可以被拆分成具有催化功能的结构域,并且这些单独表达的结构域重组在一起后可以执行完整模块的功能,虽然催化效率会有所下降[13]。在此过程中,合理选择结构的边界,保持其结构上的完整性,是获得有催化活性的结构域的关键。BacKR1的晶体结构已经被报道,根据该晶体结构确定结构域的边界,利用材料方法中的引物PCR扩增得到1 275 bp的片段,编码425个氨基酸[14]。酶切和测序验证正确的表达质粒转化到BL21 (DE3) 中,IPTG诱导表达,经镍离子亲和层析及Superdex 200凝胶过滤得到纯度>95%的目的蛋白(图3)。
2.2 外消旋聚酮类底物的还原
KR结构域的天然底物通过磷酸泛酰巯基乙胺臂连接在酰基载体蛋白 (Acyl carrier protein,ACP) 上,如图1所示,化学合成非常困难。为研究BacKR1的立体特异性,我们化学合成了聚酮底物替代物MOP-NAC。这种α位有取代基的β酮酯类化合物的α氢具有较强的酸性,手性中心很容易发生消旋化。某些KR结构域,可以特异性地选择一种底物进行不对称还原,获得4种非对映异构体中的一种[5]。还原型辅因子NADPH价格昂贵,而氧化型的NADP+相对便宜,基于来源GDH的辅因子再生系统可以有效降低反应成本[15-17]。前人的研究表明,利用上述方法可以大量制备手性纯的α-甲基-β-羟基酯作为聚酮合成的前体[11]。但并非所有的KR结构域都具有高度立体选择性,只生成一种还原产物。目前可用的KR只有A1型的AmpKR2、A2型的AmpKR1、B1型的TylKR1以及B2型的EryKR1和PikKR1[11]。
为研究BacKR1的立体选择性,利用大赛璐OC-H手性柱对其催化产物进行了分析,结果显示主要产物是(2S,3S)-构型,表明BacKR1是一种新的可用于酶法制备手性纯α-甲基-β-羟基酯聚酮合成前体的A2型KR结构域 (图4)。以α位没有甲基取代基的3-氧代戊酸-乙酰半胱胺硫酯为底物的还原研究发现超过90%的还原产物为R构型,说明α甲基对底物在BacKR1活性中心的结合有非常大的影响,导致底物以不同方式结合,从而把β酮基的不同面呈现给辅因子NADPH攻击,生成取向相反的羟基[14]。

图3 BacKR1的蛋白纯化
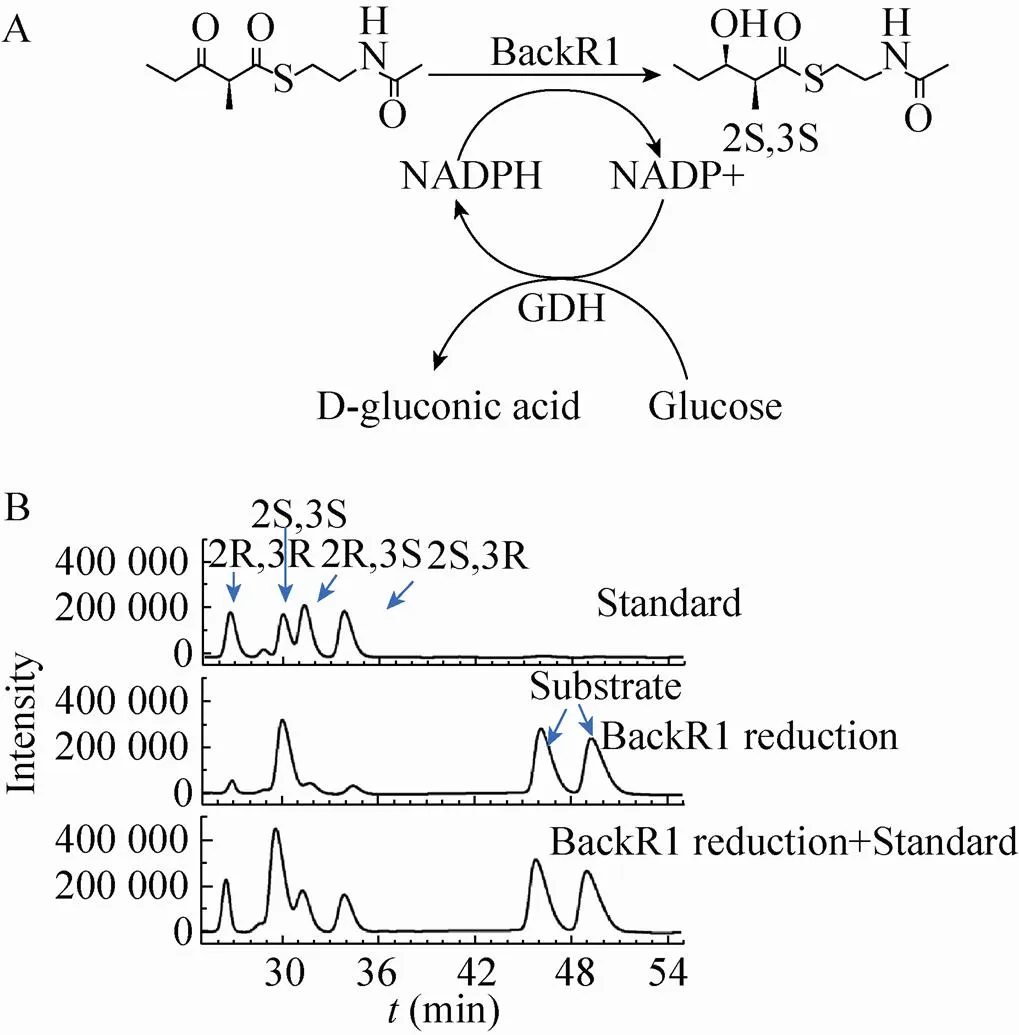
图4 BacKR1催化 (±)-2-甲基-3-氧代戊酸-乙酰半胱胺硫酯的还原
2.3 非聚酮底物的还原
除催化聚酮类底物的还原外,KR结构域还可以催化多种非聚酮底物的的还原[6-7]。在bacillaene合成过程中,BacKR1既可以还原β酮基,也可以还原α酮基,具有内在的底物宽泛性[10]。为研究其还原非聚酮底物的能力,选取了苯丙酮、苯乙酮、对氯苯乙酮、对氟苯乙酮、苯丙酮酸、环己酮、2-辛酮以及乙酰乙酸乙酯等8种化合物作为底物 (图5)。通过监测NADPH的消耗发现BacKR1可以催化环已酮和对氯苯乙酮的还原 (表1),而EryKR1则对环己酮、苯丙酮和乙酰乙酸乙酯表现出催化活性,表明二者具有不同的底物谱,各具优势[18]。

图5 本研究中采用的非天然酮基化合物

表1 通过NADPH的消耗测定聚酮BacKR1的活性
aOne unit of activity is defined as the amount of BacKR1 required to produce 1 μmol product per minute. “ND”: no detectable.
3 讨论
在聚酮类化合物生物合成过程中,KR结构域决定绝大多数甲基和羟基手性中心的立体构型[19]。通过KR结构域的替换,已经得到一些手性中心构型发生转变的新聚酮[20]。晶体结构分析揭示了KR立体选择性的分子机制,与产物手性中心构型相关的关键氨基酸残基也已经被确定,通过这些残基的突变可以改变KR的立体选择性,例如仅仅通过催化中心两个位点的突变可以把A1型KR转变成A2型[21]。然而KR结构域作为生物催化剂的潜力才刚刚被认识到。
具有多个甲基和羟基手性中心导致聚酮类化合物的全合成非常困难。利用生物催化 (包括细胞和酶) 获得手性纯的α-甲基-β-羟基酯作为聚酮合成的前体,可以大大降低聚酮合成的难度[11]。外消旋的α-甲基-β-酮酯的合成 (例如MOP-NAC) 相对简单,并且能够被KR结构域还原。但是许多单独表达的KR结构域在底物选择和酮基还原过程中的立体选择性不高,例如EryKR2还原该底物时生成所有4种可能的非对映异构体[5]。因此选择一个好的KR结构域,是α-甲基-β-羟基酯酶法制备过程中的关键。前人研究中,通过对11种不同来源KR结构域的体外分析,确定了5种不同立体选择性的KR可以用于手性纯α-甲基-β-羟基酯的大规模制备[11]。本研究中,我们发现BacKR1可以将MOP-NAC立体选择性地还原成(2S,3S)-构型产物,为A2型KR结构域在AmpKR1之外提供了另外一种选择。近期在嗜热菌中也发现了PKS,对其催化功能的研究有可能发现更稳定、效率更高的KR结构域。
手性醇作为可以引入手性中心的模块,在药物、精细化学品和农用化学品的合成中有十分广泛的应用[22-23]。酮基化合物酶法不对称还原合成手性醇因具有立体选择性强、转化效率高、生产过程对环境友好等优势越来越受到人们的重视[24-25]。目前,酵母是酮基还原酶的最重要来源[26]。然而前人的研究和本研究都显示聚酮合酶中的KR结构域可以催化多种酮基化合物的还原,并且不同来源的KR具有不同的底物谱[6-7,15]。由于聚酮化合物种类繁多,结构多样,不同来源KR所识别的底物结构差别非常大,对聚酮合酶KR结构域底物特异性的深入探索有可能发现具有应用潜力的生物催化剂。
[1] Cragg GM, Grothaus PG, Newman DJ, et al. Impact of natural products on developing new anti-cancer agents. Chem Rev, 2009, 109(7): 3012–3043.
[2] Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol, 2005, 3(12): 925–936.
[3] Cheng YQ, Coughlin JM, Lim SK, et al. Type I polyketide synthases that require discrete acyltransferases. Methods Enzymol, 2009, 459: 165–186.
[4] Zheng J, Keatinge-Clay A. The status of type I polyketide synthase ketoreductases. Med Chem Commun, 2013, 4: 34–40.
[5] Siskos AP, BaergaOA,Bali S, et al. Molecular basis of Celmer's rules: stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases. Chem Biol, 2005, 12(10): 1145–1153.
[6] Bali S, Kira J, Weissman KJ. Ketoreduction in mycolactone biosynthesis: insight into substrate specificity and stereocontrol from studies of discrete ketoreductase domains. Chembiochem, 2006, 7(12): 1935–1942.
[7] Bali S, O'Hare HM, Weissman KJ, et al. Broad substrate specificity of ketoreductases derived from modular polyketide synthases. Chembiochem, 2006, 7(3): 478–484.
[8] Calderone CT, Bumpus SB, Kelleher NL, et al. A ketoreductase domain in the PksJ protein of the bacillaene assembly line carries out both α-and β-ketone reduction during chain growth. Proc Natl Acad SciUSA, 2008, 105(35): 12809–12814.
[9] Patel PS, Huang S, Fisher S, et al. Bacillaene, a novel inhibitor of procaryotic protein synthesis produced by: production, taxonomy, isolation, physico-chemical characterization and biological activity. J Antibiot, 1995, 48(9): 997–1003.
[10] Calderone CT, Bumpus SB, Kelleher NL, et al. A ketoreductase domain in the PksJ protein of the bacillaene assembly line carries out both alpha- and beta-ketone reduction during chain growth. Proc Natl Acad Sci USA, 2008, 105(35): 12809–12814.
[11] Piasecki SK,Taylor CA, Detelich JF, et al. Employing modular polyketide synthase ketoreductases as biocatalysts in the preparative chemoenzymatic syntheses of diketide chiral building blocks. Chem Biol, 2011, 18(10): 1331–1340.
[12] Keatinge-clay A. The structures of type I polyketide synthases. Nat Prod Rep, 2012, 29(10): 1050–1073.
[13] Chen AY, Cane DE, Khosla C. Structure-based dissociation of a type I polyketide synthase module. Chem Biol, 2007, 14(7): 784–792.
[14] Piasecki SK, Zheng JT, Axelrod AJ, et al. Structural and functional studies of a trans-acyltransferase polyketide assembly line enzyme that catalyzes stereoselective alpha- and beta-ketoreduction. Proteins, 2014, 82: 2067–2077.
[15] van der Donk, WA, Zhao, H. Recent developments in pyridine nucleotide regeneration. Curr Opin Biotechnol, 2003, 14(4): 421–426.
[16] Betancor L, Berne C, Luckarift HR, et al. Coimmobilization of a redox enzyme and aco fact or regeneration system. Chem Commun, 2006, (34):3640–3642.
[17] Wallner SR, Lavandera I, Mayer SF, et al. Stereoselective anti-prelog reduction of ketones by whole cells ofin a ‘substrate-coupled’approach. J Mol Catal B Enzym, 2008, 55(3): 126–129.
[18] Li LL, Lv ZS, Guan HY, et al. Application of ketoreductase domain in polyketide synthase fromto carbonyl reduction. Huazhong Univ Sci Tech, 2011, 2(39): 72–75 (in Chinese).李凌凌, 吕早生, 关海燕, 等. 糖多孢红霉菌酮还原酶域在羰基还原中的应用. 华中科技大学学报: 自然科学版, 2011, 2(39): 72–75.
[19] Castonguay R, HeWG,ChenAY, et al.Stereospecificity of ketoreductase domains of the 6-deoxyerythronolide B synthase. J Am Chem Soc, 2007, 129(44): 13758–13769.
[20] McDaniel R, Thamchaipenet A, Gustafsson C, et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel "unnatural" natural products. Proc Natl Acad Sci USA, 1999, 96(5): 1846–1851.
[21] Zheng J, Piasecki SK, Keatinge-Clay AT. Structural studies of an A2-type modular polyketide synthase ketoreductase reveal features controlling alpha-substituent stereochemistry. ACS Chem Biol, 2013, 8(9): 1964–1971.
[22] Huisman GW, Liang J, Krebber A. Practical chiral alcohol manufacture using ketoreductases. Curr Opin Chem Biol, 2010, 14(2): 122–129.
[23] Ogawa, J, Shimizu, S. Microbial enzymes: new industrial applications from traditional screening methods. Trends Biotechnol, 1999, 17(1): 13–20.
[24] Stewart, JD. Organic transformations catalyzed by engineered yeast cells and related systems. Curr Opin Biotechnol, 2000, 11(4): 363–368.
[25] Nakamura, K, Matsuda, T. Biocatalytic reduction of carbonyl groups. Curr Org Chem, 2006, 10(11): 1217–1246.
[26] Kaluzna IA, Matsuda T, Sewell AK, et al. Systematic investigation ofenzymes catalyzing carbonyl reductions. J Am Chem Soc, 2004, 126(40): 12827–12832.
(本文责编 郝丽芳)
Heterologous expression and substrate specificity of ketoreductase domain in bacillaene polyketide synthase
Xiaohui Sun1,2*, Chengchuan Che1*, Junjie Ji2, Jianting Zheng2, and Ge Yang1
1 Collage of Life Sciences, Qufu Normal University, Qufu 273156, Shandong, China 2 State Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
The ketoreductase (KR) domain in the first extending module of the polyketide synthase (PKS) catalyzes the reductions of both ana-keto group and ab-keto group in the biosynthesis of bacillaene, suggesting the intrinsic substrate promiscuity. In order to further investigate the substrate specificity, the KR domain (BacKR1) was heterologously overexpressed in.enzymatic analysis showed that only one of the four diastereomers was formed in the reduction of the racemic (±)-2-methyl-3-oxopentanoyl--acetylcysteamine thioester catalyzed by BacKR1. In addition, BacKR1 was revealed to catalyze the reductions of cyclohexanone and p-chloroacetophenone, indicating the potential of KR domians of PKSs as biocatalysts.
polyketide, ketoreductase, heterologous expression, stereoselectivity, substrate specificity
10.13345/j.cjb.140580
November 24, 2014; Accepted:April 28, 2015
National Basic Research Program of China (973 Program) (No. 2013CB734000), National Natural Science Foundation of China (Nos. 31370101, 31400051), Beijing Municipal Natural Science Foundation (No. 5144031), the Recruitment Program of Global Youth Expertsand Initial Funds of Chinese Academy of Sciences.
Ge Yang. Tel: +86-537-4456179, E-mail:yangge100@126.com Jianting Zheng. Tel: +86-10-82545065, E-mail: jtzheng@ipe.ac.cn
*These authors contributed equally to this study.
国家重点基础研究发展计划 (973计划) (No. 2013CB734000),国家自然科学基金 (Nos. 31370101,31400051),北京市自然科学基金 (No. 5144031),青年千人计划和中国科学院启动资金资助。
2015-06-08
http://www.cnki.net/kcms/detail/11.1998.Q.20150608.1510.007.html
