柱前衍生-超高效液相色谱-串联质谱法同时检测茶叶中草甘膦和草铵膦的残留量
2015-12-26吴晓刚陈孝权肖海军刘彬球
吴晓刚 , 陈孝权, 肖海军, 刘彬球
(大益集团勐海茶业有限责任公司技术中心实验室,云南 勐海666200)
茶叶作为我国出口的主要经济作物之一,被越来越多国家的消费者所接受。目前,茶叶中农药残留问题已引起广泛关注[1]。草甘膦(glyphosate(GLY),又名镇草宁、农达等,CAS 号为1071-83-6,化学式为C3H8NO5P)[2],1970 年由孟山都公司的化学家约翰·E·弗朗茨发现。它具有内吸性,可被植物茎叶吸收向下输导,能杀死多年生杂草的地下根茎,其主要影响植物的莽草酸途径。草铵膦(glufosinate-ammonium(GLUF),又名草铵膦铵盐,CAS 号为77182-82-2,化学式为C5H15N2O4P),由赫斯特公司开发于上世纪80 年代。它属膦酸类除草剂,采用杂草茎叶定向喷雾处理[3],可以随蒸腾作用在木质部内向上运输,也可以在韧皮部向地下部分运输,是谷氨酰胺合成抑制剂。草甘膦和草铵膦均属于广谱、非选择性、灭生性除草剂[4],被广泛施用于茶园。
欧盟和日本作为我国茶叶出口的两大重要市场,均对茶叶制定了极为苛刻的农药残留限量标准[5]。我国食品安全国家标准[6]规定茶叶中草甘膦和草铵膦的最大残留限量分别为1.0 和0.5 mg/kg。日本肯定列表制度对茶叶中草甘膦和草铵膦的最大残留限量制定了较为严格的规定,分别为1.0 和0.3 mg/kg。欧盟[7]规定茶叶中草甘膦和草铵膦的最大残留限量分别为2.0 和0.1 mg/kg。从中可看出,欧盟和日本对茶叶中草铵膦的残留限量更为严格。
草甘膦和草铵膦的结构类似,含有膦酸基、羟基、氨基,为极强的两性化合物[3,8]。实际检测过程中,气相色谱和气相色谱-串联质谱技术需将草甘膦和草铵膦转化为可气化物质,会引入过多其他试剂,操作过程相对繁琐[9,10],检测效率较低;而利用配有紫外检测器的液相色谱仪进行直接测定时,因草甘膦和草铵膦的分子结构中没有共轭双键,仅存在nπ 共轭,缺少生色基团,紫外区基本无吸收[11]。近年来发展起来的超高效液相色谱-质谱联用技术,因具有检测灵敏度高、适用范围广、分析速度快和能有效排除复杂基质产生的干扰等优点,而作为首选的最佳检测手段。液相色谱-质谱直接测定草甘膦和草铵膦时,仪器响应较低,所以文献报道常采用衍生技术检测草甘膦和草铵膦残留[12,13]。9-芴甲氧羰酰氯(FMOC-Cl)作为常用衍生剂[14,15],在硼酸盐缓冲溶液中能与草甘膦和草铵膦的水提取液较好相溶,操作过程简单,衍生产物的响应信号较强,使色谱峰更容易被检测[16]。
目前国内外利用超高效液相色谱-电喷雾串联质谱(UPLC-ESI MS/MS)同时检测茶叶中草甘膦和草铵膦残留的方法报道较少,我国目前尚无茶叶中草铵膦检测方法的国家标准,按照行业标准[17]中利用液相色谱-质谱检测茶叶中的草甘膦残留时,实验过程试剂耗用量大、操作步骤极多,尤其是使用阳离子交换柱(CAX)洗脱剂洗脱,加压旋转蒸干过程易造成草甘膦回收率不稳定。本文对草甘膦和草铵膦的UPLC-MS/MS 检测参数、衍生反应最佳条件等进行了研究,前处理采用C18固相萃取柱净化,柱前FMOC-Cl 衍生,UPLC-MS/MS 检测其残留量。
1 实验部分
1.1 材料与试剂
乙腈(色谱纯,美国Fisher 公司);甲酸(色谱纯,美国Tedia 公司);十水四硼酸钠和乙酸铵(分析纯,天津市风船化学试剂科技有限公司);FMOCCl(衍生级,纯度≥99.0%,Aladdin Chemistry Co.,Ltd.);草甘膦(100 mg/L,水)购自农业部环境质量监督检验测试中心(天津);草铵膦(100 mg/L,水)购自AccuStandard,Inc.。
1.2 仪器与设备
Acquity Ultra Performance LCTM超高效液相色谱仪(沃特世科技有限公司);Waters XEVO TQD串联四极杆质谱仪(沃特世科技有限公司);高效粉碎机(上海嘉定粮油仪器有限公司);台式超声波清洗器(上海科导超声仪器有限公司);超纯水系统(成都康宁实验专用纯水设备厂);漩涡混合器(海门其林贝尔仪器制造有限公司);C18固相萃取小柱(Dikma Technologies)。
1.3 标准溶液、硼酸钠溶液和衍生液的配制
草甘膦和草铵膦标准储备液:分别用超纯水溶解并配制成质量浓度为10 mg/L 的标准储备液,于4 ℃冰箱中避光保存。
草甘膦和草铵膦混合标准工作溶液:用超纯水将上述标准储备液逐级稀释,配成含草甘膦和草铵膦均为1 mg/L 的混合标准工作溶液。
草甘膦和草铵膦基质匹配标准溶液:用经提取、净化获得的空白茶叶基质溶液将上述标准储备液逐级稀释,配成含草甘膦和草铵膦均为1 mg/L 的混合基质工作溶液。
硼酸钠溶液:称取5.00 g 的Na2B4O7·10H2O,用超纯水溶解并定容至100 mL,配制50 g/L 的硼酸钠溶液。
FMOC-Cl 丙酮溶液:称取2.00 g FMOC-Cl,用丙酮溶解并定容至100 mL,配制20 g/L 的FMOCCl 丙酮溶液。
1.4 色谱-质谱条件
1.4.1 色谱条件
色谱柱:Waters Acquity UPLC®BEH C18柱(50 mm×2.1 mm,1.7 μm);色谱柱温度:40 ℃;进样室温度:15 ℃;进样量:3 μL;流速:0.3 mL/min;流动相A:称取0.154 g 无水乙酸铵溶解于适量超纯水中,加入1.0 mL 甲酸,用超纯水定容至1 L,0.22 μm 滤膜抽滤;流动相B:纯乙腈;梯度洗脱程序:0 ~0.5 min,5% A;0.5 ~2.0 min,5% A ~95% A;2.0 ~3.5 min,95%A;3.5~4.0 min,95%A~5%A。
1.4.2 质谱条件
离子化模式:ESI+;毛细管电压:2.50 kV;离子源温度:150 ℃;气体:雾化气和脱溶剂气均为氮气,碰撞气为氩气;脱溶剂气温度:400 ℃;脱溶剂气流速:800 L/h;数据采集:多反应监测(MRM);草甘膦特征离子对(锥孔电压、碰撞能量):m/z 392 >88(25 V、20 V)、m/z 392>179(25 V、10 V),其中m/z 392>88 为定量离子对;草铵膦特征离子对(锥孔电压、碰撞能量):m/z 404 >136(25 V、25 V)、m/z 404>179(25 V、10 V),其中m/z 404>136 为定量离子对。
1.5 样品前处理
采样和试样制备:选取巴达山的毛茶空白样品,用四分法缩减取200 g,粉碎后过1.0 mm 孔径筛,混匀,装入样品盒中,备用。
提取:称取均质好的试样1 g(精确至0.001 g)置于50 mL 具塞聚乙烯离心管中,加入20 mL 超纯水,充分振荡2 min,再加入二氯甲烷5 mL,混匀后超声提取30 min,以4 500 r/min 转速离心5 min,上清液待净化。
净化:取1.0 mL 上清液淋洗C18固相萃取小柱(事先分别用5 mL 甲醇、5 mL 超纯水活化),弃去流出液;再重新加入2 mL 上清液过C18固相萃取小柱,流出液放入10 mL 玻璃离心管中,待衍生。
衍生:取1.0 mL 的净化液,分别依次加入1.0 mL 50 g/L 硼酸钠缓冲溶液、1.0 mL 20 g/L FMOCCl 丙酮衍生液,混匀后室温下衍生2 h,以4 500 r/min 转速离心5 min,过0.2 μm 有机微孔滤膜后,供超高效液相色谱-串联质谱测定。
2 结果与讨论
2.1 质谱条件的优化
2.1.1 离子化模式及定性、定量离子对的选择
分别用超纯水配制1.0 mL 质量浓度均为10 mg/L 的草甘膦和草铵膦标准溶液,依次加入50 g/L 硼酸盐缓冲溶液和20 g/L 的衍生剂溶液各1.0 mL,室温下过夜衍生,将各单标准溶液放入进样器中,在梯度洗脱条件下用Waters Acquity UPLC®BEH C18柱(50 mm×2.1 mm,1.7 μm)进行分离。在正离子模式下进行MS2扫描,采集范围为m/z 350~450,得到草甘膦和草铵膦的母离子峰;分别对草甘膦和草铵膦衍生物进行子离子扫描,分别输入已找到的母离子参数,子离子采集范围为m/z 60 ~410,调节碰撞能量,以获得稳定性好、信号强度高的离子碎片。

图1 正离子模式下GLY-FMOC 和GLUF-FMOC 的MS2 和子离子扫描光谱图Fig.1 MS2 and daughter scan spectra of GLY-FMOC and GLUF-FMOC in positive ion mode
正离子模式下,GLY-FMOC 和GLUF-FMOC 的MS2和子离子扫描光谱如图1 所示。锥孔电压20 V 下,GLY-FMOC 和GLUF-FMOC 的MS2扫描中分别显示m/z 392 和m/z 404 母离子峰;碰撞能量20 V 下,GLY-FMOC 子离子扫描显示m/z 88、m/z 170、m/z 179 和m/z 214 等离子碎片,GLUFFMOC 子离子扫描显示m/z 136、m/z 182、m/z 179 和m/z 208 等离子碎片。参考文献[16],选择响应较高的m/z 392>88、m/z 392>179 和m/z 404>136、m/z 404>179 分别作为草甘膦和草铵膦衍生产物的定量、定性离子对。
2.1.2 质谱参数的优化
通过手动调节毛细管电压、脱溶剂气温度、锥孔电压、碰撞能量和流速等,在MRM 模式下,考察不同参数下草甘膦和草铵膦衍生物离子的信号响应强度,从而确定其最佳质谱采集参数。最终,选取如1.4 节所示质谱参数。
2.2 衍生途径及母离子裂解途径分析
根据2.1 节中考察得到的各母离子及子离子参数,对GLY-FMOC 和GLUF-FMOC 衍生途径及其裂解后的子离子产生途径进行分析。图2a 显示GLY 和GLUF 与FMOC-Cl 衍生的过程,期间GLY和GLUF 的N-H 键和FMOC-Cl 的C-Cl 键断裂,脱去HCl。图2b 显示GLY-FMOC 和GLUF-FMOC均可以脱去一个CO2及其衍生前的GLY 和GLUF,而生成一个共有的m/z 179 的结构;GLY-FMOC 和GLUF-FMOC 也可以脱去一个m/z 222 结构,生成GLY 和GLUF 加H+产物。图2c 显示GLY-FMOC中间产物m/z 170 裂解生成m/z 88;GLUF-FMOC裂解生成m/z 208,其中间产物m/z 182 裂解生成m/z 136。
2.3 色谱条件的优化
2.3.1 色谱柱的选择
Waters Acquity UPLC®BEH C18柱(50 mm×2.1 mm,1.7 μm)采用1.7 μm 小颗粒填料在更宽的线速度范围内可获得最佳的柱效,能显著提升色谱柱的性能[18]。众多研究型文献对利用C18色谱柱分离草甘膦和草铵膦均有较全面的报道。林永辉等[3]曾利用Kinetex C18色谱柱对茶叶中的草铵膦进行分离、确证(柱前衍生),平均回收率为61.6%~81.4%,LOD 值为0.03 mg/kg;曹赵云等[19]曾利用Zorbax Extend C18色谱柱对稻米中的草甘膦进行定性、定量分析(柱前衍生),平均回收率为85.0%~113.6%,LOD 值为0.002 mg/kg。
2.3.2 流动相的优化
配制以下4 组流动相:(Ⅰ)A 为乙腈,B 为0.154 g 无水乙酸铵溶于1 L 超纯水中,其中加入1 mL 甲酸;(Ⅱ)A 为乙腈,B 为0.1%(v/v)甲酸水溶液;(Ⅲ)A 为50%(v/v)甲醇乙腈,B 为0.1%(v/v)甲酸水溶液;(Ⅳ)A 为甲醇,B 为0.1%(v/v)甲酸水溶液。考察草甘膦和草铵膦在使用上述各组流动相梯度洗脱条件下的色谱行为。实验结果显示:流动相Ⅰ出峰效果最好;Ⅱ中分析物有明显的峰拖尾现象;含有甲醇的流动相(Ⅲ和Ⅳ)出峰时间延后。故本实验选取流动相Ⅰ,其流动相中的甲酸可提供H+,能维持目标离子质子化状态,增强目标分析物在流动相中的离子化程度,有助于提高检测效率;铵盐可以改善样品峰形,使峰形更对称。
2.4 衍生条件的优化
配制质量浓度为20、30、40、50、60 g/L 的硼酸钠缓冲溶液,及质量浓度为1、5、10、20、30 g/L 的FMOC-Cl 丙酮溶液,做二者对草甘膦和草铵膦的衍生效果影响试验。当硼酸钠缓冲溶液质量浓度为50 g/L 和FMOC-Cl 丙酮溶液质量浓度为20 g/L时,衍生效果最佳。在50 g/L 硼酸钠缓冲溶液和20 g/L FMOC-Cl 丙酮溶液下,进行衍生时间分别为1、1.5、2、2.5、3、3.5、4、4.5、5 h 的条件试验,发现衍生时间为2 h 时,衍生反应已达到平衡。
2.5 方法检出限及基质效应评定
基质效应(matrix effect,ME)主要来源于色谱分离过程中与待测物共流出的干扰物质(包括内源性组分和外源性组分)对待测物离子化效率的影响。内源性组分是指茶叶样品提取过程中被同时提取出来的有机或无机分子(如无机盐、酚类、色素和脂类等)。茶叶中复杂的内源性基质因茶种类的不同而有所区别,例如绿茶、红茶、乌龙茶和普洱茶[20]。而外源性组分主要是由前处理过程引入,包括衍生反应时引入的缓冲盐、衍生剂及其溶解液等。
为分析茶叶内源基质和衍生反应对草甘膦和草铵膦产生的基质效应,准备如下两组实验:(A)将草甘膦和草铵膦用超纯水配制成质量浓度为0.009 375、0.018 75、0.075、0.15 和0.3 mg/L 的混合标准溶液。每个浓度样品取1.0 mL,依次加入1.0 mL 50 g/L 硼酸钠缓冲溶液和1.0 mL 20 g/L衍生剂溶液,衍生2 h;(B)将草甘膦和草铵膦用空白基质液稀释成0.009 375、0.018 75、0.075、0.15和0.3 mg/L 的混合标准溶液。每个浓度样品取1.0 mL,依次加入1.0 mL 50 g/L 硼酸钠缓冲溶液和1.0 mL 20 g/L 衍生剂溶液,衍生2 h 后进样分析。以质量浓度(X,mg/L)为横坐标,定量离子峰面积(Y)为纵坐标建立标准曲线。由表1 可见,各标准曲线相关系数(r)均大于0.990,相关性良好。在信噪比(S/N)为3 时计算检出限(LOD),S/N 为10 时计算定量限(LOQ),结果见表1。0.025 mg/L GLY-FMOC 和GLUF-FMOC 混合纯水标准溶液和基质匹配标准溶液的色谱图见图3。

图2 (a)GLY 和GLUF 的化学结构及其衍生反应过程、(b)GLY-FMOC 和GLUF-FMOC 的共有裂解途径和(c)GLY-FMOC 和GLUF-FMOC 各自特有的裂解途径Fig.2 (a)Chemical structures of GLY and GLUF,and derivatization reaction with FMOC-Cl,(b)common fragmentation pathway for the two derivatives and (c)specific fragmentation pathways for GLU-FMOC and GLUF-FMOC
基质效应可以用ME =B/A×100%计算,式中B为基质曲线斜率,A 为标准曲线斜率。一般情况下,ME 在85%~115%之间不存在明显的基质效应。据此判断,本检测方法对草甘膦和草铵膦基质效应不显著(见表1)。
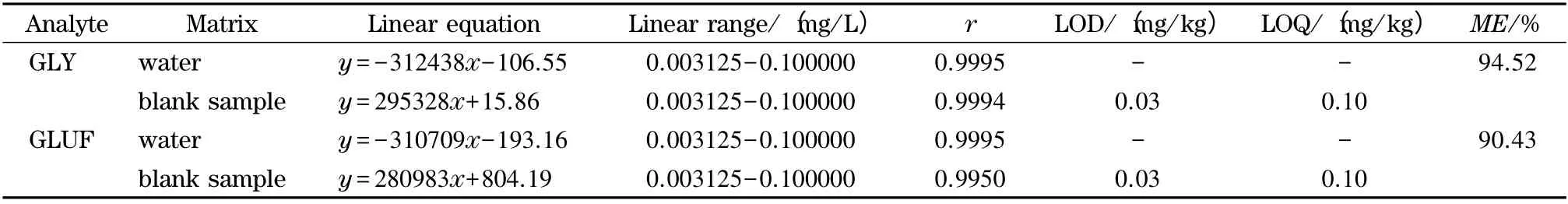
表1 草甘膦和草铵膦标准溶液和基质匹配标准溶液的线性方程、相关系数和基质效应(ME)Table 1 Linear equations,correlation coefficients (r)and matrix effects (ME)of GLY and GLUF standard solutions and matrix-matched standard solutions

图3 GLY-FMOC 和GLUF-FMOC 标准溶液(0.025 mg/L)的色谱图Fig.3 Chromatograms of GLY-FMOC and GLUF-FMOC standard solutions at 0.025 mg/L
2.6 方法的回收率、精密度及其稳定性
准确称取1 g 匀质好的空白茶样于50 mL 离心管中,分别添加草甘膦和草铵膦各0.375、1.500 和4.500 mg/kg,每个添加浓度重复6 次,按照1.5 节所述进行处理后进样。由表2 可见,3 个添加水平下,草甘膦平均回收率为87.37%~99.11%,相对标准偏差(RSD)(n =6)为0.68%~1.35%;草铵膦平均回收率为81.44% ~86.17%,相对标准偏差(RSD)(n =6)为1.01%~2.33%。取一份处理好的加标1.500 mg/kg 溶液,在进样室内放置,每隔4 h测定一次,结果测得草甘膦和草铵膦定量离子峰面积的RSD 分别为2.65%和4.01%,表明供试品溶液在24 h 内稳定。

表2 茶叶中2 种农药的添加回收率和相对标准偏差(n=6)Table 2 Recoveries and relative standard deviations(RSDs)of the two pesticides spiked in tea(n=6)
2.7 实际样品的测定
利用本检测方法对来自云南省普洱茶主产区的39 份茶样原料、11 份市售绿茶样本和7 份市售红茶样本进行草甘膦和草铵膦残留量的测定。结果显示,草甘膦和草铵膦均未检出。
3 结语
本方法通过用超纯水和二氯甲烷作为提取溶剂较好地去除茶叶中脂溶性色素等物质,C18固相萃取小柱净化,优化UPLC-MS/MS 仪器条件,建立了柱前衍生-超高效液相色谱-串联质谱测定茶叶中草甘膦和草铵膦两种农药残留量的检测方法。本方法净化过程简单,检测速度快,可作为茶叶中草甘膦和草铵膦残留量的分析确证方法。
[1] Xu J,Chen J,Ye H Y,et al. Journal of Instrumental Analysis (徐娟,陈捷,叶弘毅,等. 分析测试学报),2011,30(9):990
[2] Mo J L,Miao L,Gan N J. Modern Food Science and Technology (莫佳琳,缪璐,干宁军. 现代食品科技),2011,27(9):1143
[3] Lin Y H,Liu Z C,Yang F,et al. Chinese Journal of Chromatography (林永辉,刘正才,杨方,等. 色谱),2012,30(12):1260
[4] Yoshioka N,Asano M,Kuse A,et al. J Chromatogr A,2011,1218(23):3675
[5] Hu B Z,Cai H J,Song W H. Chinese Journal of Chromatography (胡贝贞,蔡海江,宋伟华. 色谱),2012,30(9):889
[6] GB 2763-2014
[7] Zhu L,Chen H P,Zhou S J,et al. Chinese Journal of Analytical Chemistry (诸力,陈红平,周苏娟,等. 分析化学),2015,43(2):271
[8] Jiang Y,Cao Z Y,Jia R L,et al. Chinese Journal of Chromatography (江燕,曹赵云,贾瑞琳,等. 色谱),2012,30(1):39
[9] Hu J Y,Zhao D Y,Ning J,et al. Chinese Journal of Pesticide Science (胡继业,赵殿英,宁君,等. 农药学学报),2007,9(3):285
[10] Ma W M,Niu S,Li D Y,et al. Agrochemicals (马为民,牛森,李东运,等. 农药),2006,45(4):261
[11] Li X J,Meng P J,Wang Y Y,et al. Physical Testing and Chemical Analysis Part B:Chemical Analysis (李小娟,孟品佳,王燕燕,等. 理化检验:化学分册),2015,51(3):307
[12] Chen L. Tea Science and Technology (陈磊. 茶叶科学技术),2014(1):21
[13] Feng Y C,Ma L L,Jia L,et al. Journal of Food Safety and Quality (冯月超,马立利,贾丽,等. 食品安全质量检测学报),2014,5(4):1147
[14] Lee E A,Zimmerman L R,Bhullar B S,et al. Anal Chem,2002,74(19):4937
[15] Hidalgo C,Rios C,Hidalgo M,et al. J Chromatogr A,2004,1035(1):153
[16] Ibanez M,Pozo O J,Sancho J V,et al. J Chromatogr A,2005,1081(2):145
[17] SN/T 1923-2007
[18] An R,Bo M P. Modern Science Instruments (安蓉,薄美萍. 现代科学仪器),2006(1):20
[19] Cao Z Y,Mou R X,Chen M X. Chinese Journal of Chromatography (曹赵云,牟仁祥,陈铭学. 色谱),2010,28(8):743
[20] Chen G Q,Cao P Y,Liu R J. Food Chem,2011,125(4):1406
