木质素类型及添加量对AGM阀控式铅酸蓄电池负极性能的影响研究
2015-12-24张祖波夏诗忠戴长松
张 兴,张祖波,2,夏诗忠,戴长松
(1.湖北骆驼蓄电池研究院有限公司,湖北 襄阳 441000;2.哈尔滨工业大学化学与化工学院,黑龙江 哈尔滨 150001)
0 前言
木质素是迄今为止改善电池性能最为有效的有机添加剂之一。作为一种阴离子表面活性剂,木质素无论被吸附在 Pb 或者 PbSO4上,都会使得负极活性物质的晶体大小和形貌结构发生变化。当木质素在 Pb 晶体上选择性吸附时具有活化作用,而在PbSO4上选择性吸附时,则由于增加了 PbSO4的过饱和度,具有一定的钝化作用[1-7]。目前对木质素的选择仍然以经验居多,选择既有利于提升充电接受能力同时又能提升放电性能的的木质素是极其困难的。本文则以铅酸蓄电池行业内三种主流的木质素作为研究对象,对不同类型、不同添加量木质素对AGM 阀控式铅酸蓄电池负极性能的影响趋势进行详细地研究论证。
1 实验
1.1 仪器与试剂
Digatron BTS-600 电池测试系统(迪卡龙青岛电子科技有限公司),GSL-101BI 激光颗粒分布测量仪(辽宁仪表研究所有限公司),BRUKER 傅里叶红外光谱仪(德国布鲁克仪器有限公司),BTS-5V20A 新威电池检测系统(深圳市新威尔电子有限公司),WD4005S 高低温试验箱(上海建恒仪器有限公司),恒温恒湿固化箱(上海建恒仪器有限公司),精密可调恒温水浴槽(杭州九环环境试验设备有限公司),3 Ah AGM 阀控式单体铅酸蓄电池(自制),木质素 A(外购),木质素 B(外购),木质素 C(外购),无水乙醇(上海国药试剂有限公司)等。
1.2 电池制备
实验电池为 3 Ah AGM 阀控单体铅酸蓄电池(三正两负极群组结构,负极限制容量,其中负极板配方中含不同类型、不同添加量木质素,负极板采用中温高湿固化工艺),AGM 隔板隔离正负极板,其中 AGM 隔板双包负极,采用内化成工艺,化成电解液为ω(Na2SO4)=1.1%且ρ=1.260 g/cm3的硫酸溶液。
1.3 性能测试
1.3.1 充电接受能力测试
本部分性能测试参照 GB/T 5008.1-2013 要求,具体如下:蓄电池充满电后 1~3 h 内以I10=C20/10恒流放电 5 h,然后在 0 ℃ 温度下静置 24 h,取出电池,在 2 min 内以 2.4 V 恒压充电 10 min,记录第 10 min 的充电电流ICa,并且根据电流–时间的变化曲线进行积分计算出 10 min 内实际充入的电量Q10min,并计算ICa/I10比值以及Q10min/C20比值。
1.3.2 -18 ℃ 低温高倍率放电性能测试
蓄电池充满电之后,转入 -18 ℃ 低温箱中静置24 h,然后取出电池,在 2 min 内以Icc=160I20进行放电直至电池端电压小于 1.2 V 为止,记录第 30 s放电电压并记录放电至电池端电压小于 1.2 V 持续放电时间t,并根据电流–时间变化曲线进行积分,计算放电至电池端电压小于 1.2 V 时电池在 -18 ℃低温条件下放电容量Ce与C20的比值。
1.3.3 活性物质利用率测试
1.3.3.1 20 小时率活性物质利用率测试
蓄电池充满电之后,以I20放电至 1.75 V,记录放电时间t并根据电流–时间曲线进行积分计算20 小时率放电容量C20以及 20 小时率活性物质利用率η20。其中η20=C20÷6.0043×100%。
1.3.3.2 1小时率活性物质利用率测试
蓄电池充满电之后,以I1放电至 1.6 V,记录放电时间t并根据电流–时间曲线进行积分计算 1小时率放电容量C1以及 1 小时率活性物质利用率η1。其中η1=C1÷6.0043×100%。
1.3.4 Peukert 曲线测试(倍率放电性能测试)
蓄电池充满电之后,测试 0.2C20、0.3C20、0.5C20、0.7C20、1C20、1.5C20、2C20常温倍率放电性能,记录电池达到规定放电电压终止条件时的放电时间t,其中规定: 0.2C20、0.3C20放电终止电压为 1.75 V,0.5C20、0.7C20放电终止电压为 1.67 V,1C20、1.5C20、2C20放电终止电压为 1.6 V。
1.3.5 HRPSoC 循环寿命测试
电池充满电后 1~2 h 内以I=2I20恒流放电 5 h至 50%荷电状态,终止条件为 1.75 V,然后做如下循环:以I=3I1恒流充电 1 min(注:I1为 1 小时率放电电流);静置 1 min 后,再以I=3I1恒流放电 1 min ;静置 1 min。循环过程中,充电电压高于 2.83 V 或放电电压低于 1.73 V 即达到寿命终止条件。
2 结果与讨论
2.1 材料表征
2.1.1 粒径分布测试

图1 不同类型木质素粒径分布
从粒径分布图1中可以看出:木质素 A 粒径分布非常不均一,主要集中在 0.5~1.0 μm 和 1.0~2.0 μm 范围内,其比例分别为 18.86%和 18.94%,而粒径超过 5 μm 以上的,木质素 A 也占据了近 40%,而木质素 B 的粒径分布非常均匀,主要集中分布在 0.5~1.0 μm、1.0~2.0 μm 和 2.0~5.0 μm,其比例分别为 24.21%、30.98%和 31.52%,粒径超过 5 μm 以上的分级比例远远低于木质素 A,说明木质素 B 平均粒径分布比木质素 A 更加均匀集中,平均颗粒尺寸也比木质素 A 更小。
2.1.2 红外光谱测试
在图2中可以清晰看出,峰 1 为羟基 -OH 的特征峰,波数 3500 cm-1,峰 2 为 -CH3的收缩振动峰,峰 3 为 C=O 的特征峰,峰形尖锐。在 1300~800 cm-1指纹区域内,除单键的伸缩振动外,还有因变形振动产生的谱带。这种振动与整个分子的结构有关,当分子结构稍有不同时,该区的吸收就有细微的差异,在 900~600 cm-1区域内,木质素 C出现弱吸收峰带,这可能是由于分子中存在两个以上 -CH2- 组成的长链或环状链。
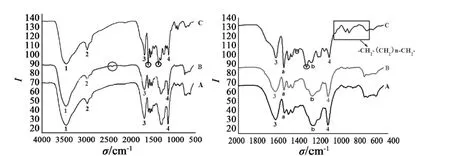
图2 不同类型木质素红外光谱
2.2 电池性能测试
2.2.1 活性物质利用率
从图3中可以看出,随木质素 A、B 在活物质中所占质量分数的增加,20 小时率活性物质利用率和 1 小时率活性物质利用率都逐渐降低;而随木质素 C 所占质量分数的增加,20 小时率活性物质利用率呈抛物线状上升—下降趋势,当其质量分数为 0.2%时,20 小时率活性物质利用率和 1 小时率活性物质利用率达到最大值;当质量分数在0.1%~0.3%范围内,采用木质素 C 作为有机膨胀剂时负极活性物质利用率比用木质素 A、木质素 B为膨胀剂时偏低,这可能是木质素 C 的结构和在硫酸溶液中溶解性与木质素 A、木质素 B 不同造成的。
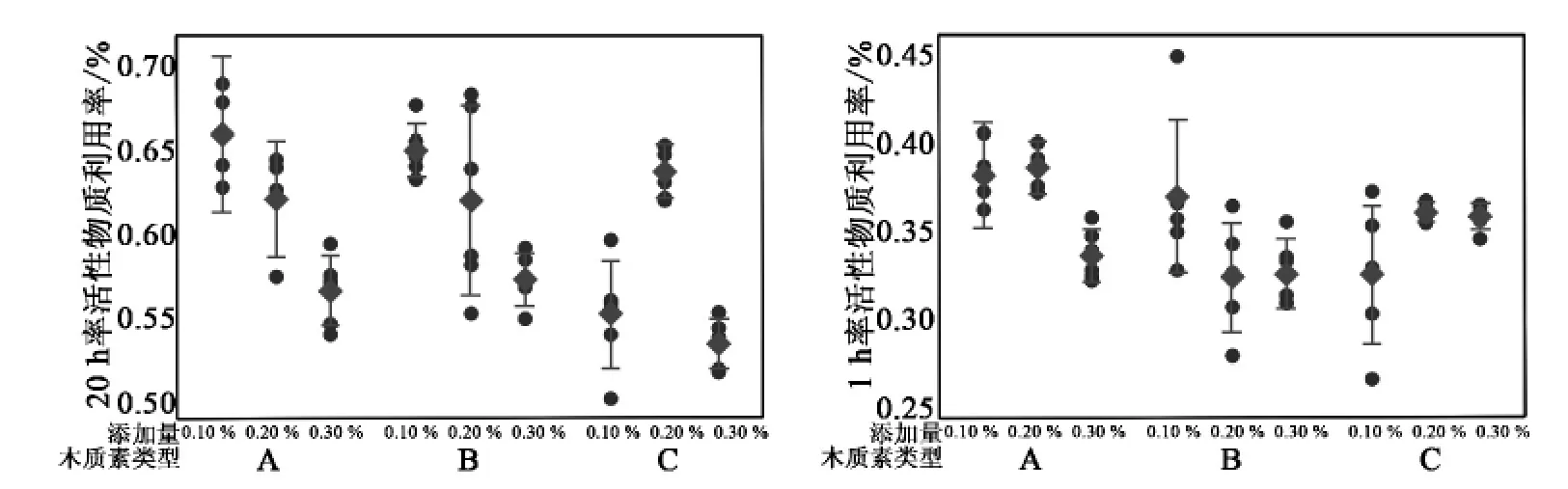
图3 木质素类型和质量分数不同时活性物质利用率的区间趋势图
当分别取 200 mg 上述三种木质素溶解于ρ=1.28 g/mL 的硫酸溶液中,如图4所示,发现,木质素 A 和木质素 B 在硫酸溶液中的溶解度较大,但是木质素 C 的溶解度较小,说明木质素 A 和木质素 B 相对于木质素 C 具有较多的亲水性基团,这样将三种木质素作为有机添加剂加入到负极活性物质中,由于木质素的溶解–造孔作用,可能会提高负极活性物质的孔隙率及孔径,高孔隙率更有利于硫酸的渗透和扩散作用[8],而活性物质利用率主要受硫酸浓度和扩散速度控制,因此采用木质素A、木质素 B 作为有机膨胀剂,同时采用内化成工艺时,更有利于提高负极活性物质的利用率。

图4 木质素在硫酸水溶液中分散性
2.2.2 充电接受能力
从图5中可以看出,随木质素 A、C 在活物质中所占质量分数的增加,充电接受能力呈抛物线状上升—下降趋势,而随着木质素 B 在活性物质中所占质量分数的增加,充电接受能力呈直线逐渐降低。由于负极充电是溶解—电化学还原过程,因此硫酸铅的溶解度将直接限制负极充电过程是否顺利进行[9]。当木质素在活物质中所占质量分数处于一定范围内时,木质素的加入可能会使硫酸铅很容易吸附在添加剂质点上,有利于防止硫酸铅钝化层的形成[10-12],降低体系的表面能[9],在一定程度上促进了硫酸铅的溶解,进而提高了负极的充电接受能力[5,13],但当所添加木质素的质量分数超过 0.2%时,充电接受能力均会出现不同程度的下降,这可能是由于过多木质素的加入使得负极活性物质的导电性有所下降[14],由于木质素类型不同,其所含亲水性、憎水性基团数量和质均分子量分布存在差异,对负极充电接受能力的影响会有很大差异。木质素 A、B 属于同系列阴离子表面活性剂,但 A、B 两种木质素的质均分子量有差异,木质素 B 的质均分子量比木质素 A 的质均分子量大,说明木质素的平均分子量分布对充电接受能力有一定影响。
2.2.3 -18℃ 低温起动性能
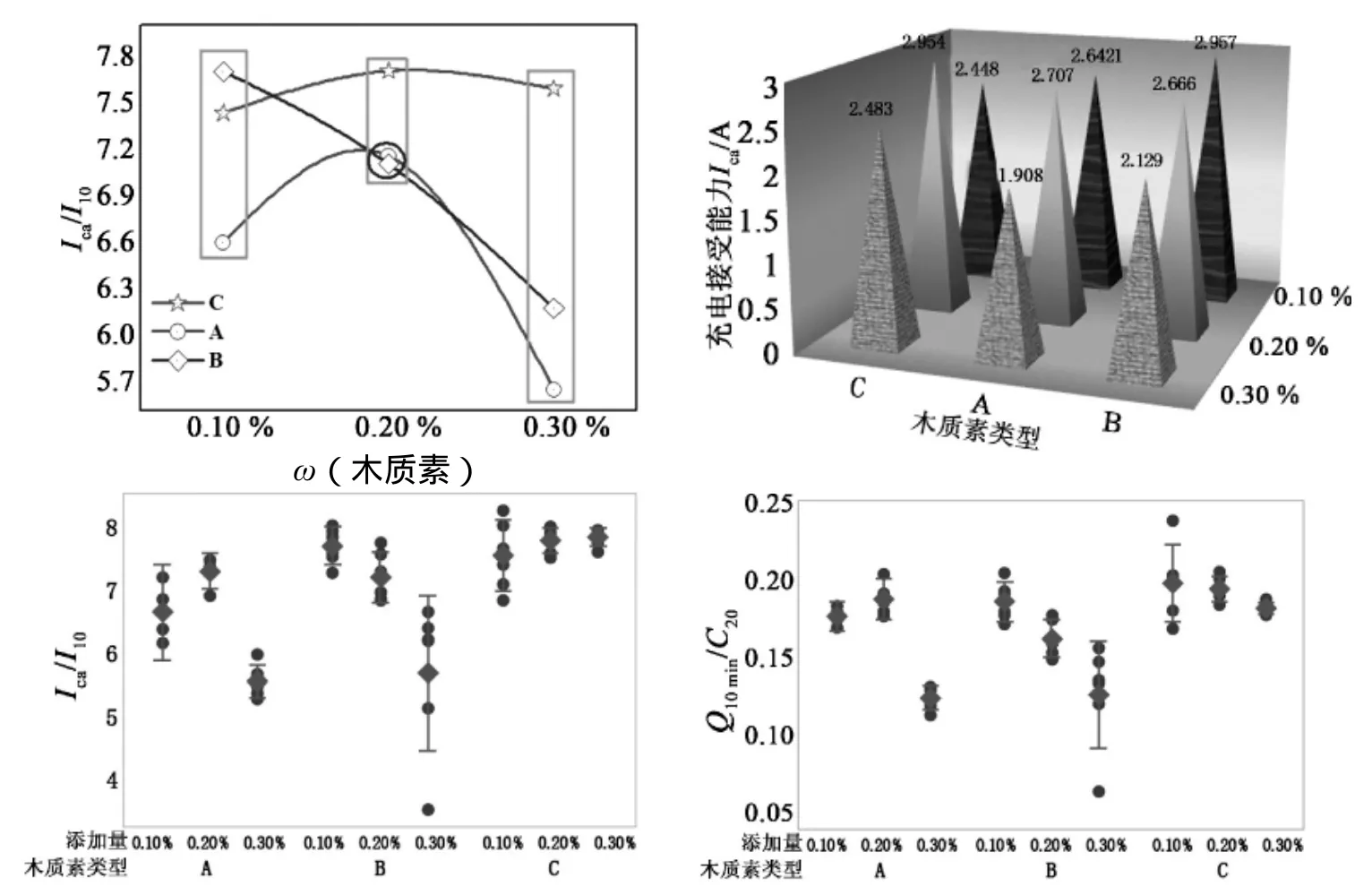
图5 不同类型、不同添加量木质素充电接受能力
从图6可以看出,随木质素 A、C 在活物质中所占质量分数的增加,-18 ℃ 低温高倍率放电性能逐渐增强,但随着木质素 B 在活性物质中所占质量分数的增加,-18 ℃ 低温高倍率放电性能呈抛物线状上升—下降趋势。当木质素在活性物质中所占质量分数在 0.1%~0.3%范围内时,随着质量分数的增加,采用木质素 A、木质素 C 为有机膨胀剂的负极在 -18 ℃ 低温高倍率下的放电性能逐渐增强,但当质量分数超过 0.2%时,采用木质素 B 作为有机膨胀剂的负极在 -18 ℃ 低温高倍率下的放电性能反而有所降低。
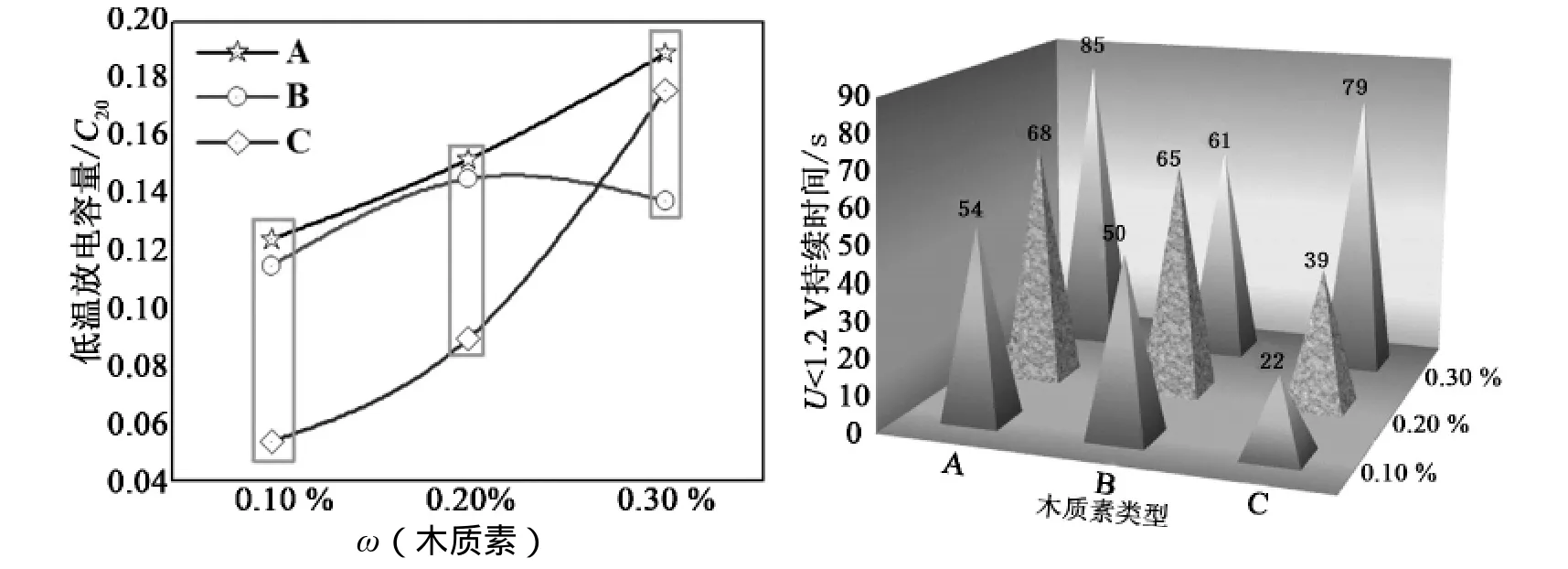
图6 木质素类型和质量分数不同时 -18 ℃ 低温起动性能
2.2.4 倍率放电性能(Peukert 曲线测试)
当木质素的质量分数不同时,通过测试放电倍率条件下电流–时间关系曲线,并对曲线进行拟合发现,如图7所示,在 0.2C20~2C20范围内电流–时间关系曲线能较好地符合 Peukert 方程t=K/In关系(R2>99%),其中随着木质素所占质量分数的增加,木质素 A 的n值逐渐增大,而木质素 B 和木质素 C 的n值则逐渐减小,刚好相反,说明当所占质量分数相同时,采用木质素 A 作为有机添加剂时,对负极不同倍率放电性能的影响要比采用木质素 B、木质素 C 时大。
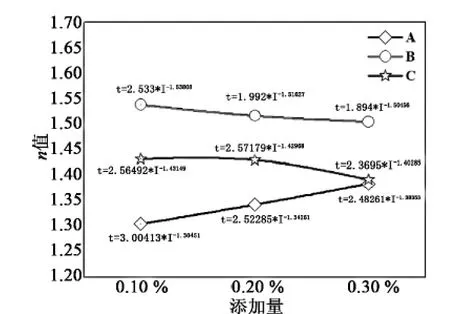
图7 木质素类型和质量分数不同时不同倍率放电性能
2.2.5 HRPSoC 循环寿命
从图8中可以看出,随着木质素所占质量分数的增加,高倍率循环寿命均缩短了。这是由于部分荷电状态下高倍率充放电过程中,负极板出现了强烈的硫酸盐化,而随着负极板中木质素质量分数的增加,大量的木质素分子质点吸附在负极活性物质表面[2],降低了负极实际参与电化学反应的面积[4],负极极化内阻增加,加剧了负极活性物质硫酸盐化程度[7],在高倍率短时间放电时,导致负极表面形成致密而细小的硫酸盐层[1],而 60 s 静置时间会促使硫酸铅晶体的长大,继而在进行高倍率恒流充电时,降低了负极析氢过电位[15],造成负极提前剧烈析氢,导致在循环过程中负极的动态充电效率不断降低。但在同一添加量下进行比较时,采用木质素 C 时高倍率循环寿命略长。尤其当木质素C 所占质量分数达到 0.1%时,循环寿命达到 6740次,远远高于采用木质素 A 或木质素 B 时,这是由于此时负极活性物质高倍率下的充电电压显著降低,电化学极化程度较低。
3 结论
(1)采用内化成工艺时,采用木质素 A 或木质素 B 作为有机膨胀剂,更有利于提高负极活性物质利用率。
(2)当添加的木质素在活性物质中所占质量分数超过 0.1%时,推荐采用木质素 C 作为有机膨胀剂,因为这样负极充电接受能力较好。
图8 木质素类型和质量分数不同时部分荷电状态下高倍率循环寿命
(3)随着木质素在活性物质中所占质量分数的增加,负极在部分荷电状态下的高倍率循环寿命逐渐降低,当采用木质素 C 作为有机膨胀剂且其所占质量分数为 0.1%时,负极在部分荷电状态下的高倍率循环寿命最佳。
(4)当木质素在活性物质中所占质量分数在0.1%~0.3%范围内,随着木质素所占质量分数的增加,负极在 -18 ℃ 低温高倍率放电性能逐渐增强,但采用木质素 B 作为有机膨胀剂时,当其质量分数超过 0.2%时,-18 ℃ 低温高倍率放电性能反而有所降低,所以推荐采用木质素 A,用其为有机膨胀剂的负极 -18 ℃ 低温高倍率放电性能较好。
(5)为满足 AGM 起停电池对高充电接受能力和长寿命的需求,推荐将 A 与 C 进行混合添加,具体将在今后的研究中进一步完善。
[1] Sawai K, Funato T, Watanabe M, et al.Development of additives in negative active-material to suppress sulfation during high-rate partial-state-of-charge operation of lead–acid batteries[J].Journal of Power Sources.2006, 158(2): 1084-1090.
[2] Pavlov D, Myrvold B O, Rogachev T, et al.A new generation of highly efficient expander products and correlation between their chemical composition and the performance of the lead–acid battery[J].Journal of Power Sources.2000, 85(1): 79-91.
[3] Hirai N, Tanaka T, Kubo S, et al.Density and hardness of negative pastes of lead–acid batteries containing organic additives with or without quinone structure[J].Journal of Power Sources.2006, 158(2): 1106-1109.
[4] Sawai K, Funato T, Watanabe M, et al.Development of additives in negative active-material to suppress sulfation during high-rate partial-state-of-charge operation of lead–acid batteries[J].Journal of Power Sources.2006, 158(2): 1084-1090.
[5] Prengaman R D.Improvements to active material for VRLA batteries[J].Journal of Power Sources.2005, 144(2): 426-437.
[6] Matrakova M, Rogachev T, Pavlov D, et al.Influence of phenolic group content in lignin expanders on the performance of negative lead–acid battery plates[J].Journal of Power Sources.2003, 113(2): 345-354.
[7] Papazov G, Pavlov D, Monahov B.Influence of temperature on expander stability and on the cycle life of negative plates[J].Journal of Power Sources.2003, 113(2): 335-344.
[8] Boden D P, Arias J, Fleming F A.The effect of organic expander materials on the performance,life, surface area and crystal structure of negative electrodes in valve regulated cells[J].Journal of Power Sources.2001, 95(1–2): 277-292.
[9] 李建华,孙杰英.关于负极膨胀剂及负极有机膨胀剂(一)[J].蓄电池.2004(04): 151-154.
[10] 陈小川,林永兴.铅酸蓄电池负极有机膨胀剂吸附作用的初步分析[J].蓄电池.2006(04):171-174.
[11] 陈代武,李绿冰,林目玉,等.铅酸蓄电池添加剂的研究进展[J].电源技术.2012(08):1245-1247.
[12] 敖建平,孙国忠.添加剂和极板结构对铅酸蓄电池冷起动性能的影响[J].蓄电池.1996(03):29-36.
[13] Boden D P, Arias J, Fleming F A.The effect of organic expander materials on the performance,life, surface area and crystal structure of negative electrodes in valve regulated cells[J].Journal of Power Sources.2001, 95(1–2): 277-292.
[14] Hoffmann G, Vielstich W.The influence of organic expanders on the kinetics of the lead electrode[J].Journal of Electroanalytical Chemistry and Interfacial Electrochemistry.1984, 180(1–2):565-576.
[15] Saez F, Martinez B, Marin D, et al.The influence of different negative expanders on the performance of VRLA single cells[J].Journal of Power Sources.2001, 95(1–2): 174-190.
