无菌药品GMP认证后生产现场质量管理探讨
2015-12-22李丽华
李丽华
(广西梧州制药〈集团〉股份有限公司,广西 梧州 543000)
实施药品生产质量管理规范(GMP)认证是国家对药品生产企业监督检查的一种手段,也是药品监督管理工作的重要内容。同时GMP认证后可以优化企业的生产管理队伍建设,培养大量的技术管理人员,改善落后管理模式、工作方式和生产观念。因此,生产管理建设是一项长期性任务。
1 新版GMP 关键条款解读
新版GMP 第一百三十八条至第一百四十八条里强调对验证文件管理方面的技术要求。在日常工作中,要做好验证和再验证工作的总体计划,认真开展工艺验证、公用设施和系统验证、设备验证、清洁验证等各项验证工作通过开展验证工作,保证工艺参数、操作程序、重要变量等的可靠性、稳定性,减少产品返工和复检次数,确保产品质量。
2 加强原辅包材质量的控制
原辅包材质量的好坏是决定药品质量的关键。药品的原辅包材如果发生变化,那么势必会对药品的质量造成很大的波动。因此,变更原辅包材时必须按照规定通过工艺的验证,以验证数据作支持,杜绝一切随意的变更。如确需变更,应按程序上报药品监督管理部门,使变更合法化、规范化和科学化。在验证的过程中,可以随时根据具体条件调整工艺参数,直到验证结束。经验证后的结果应及时编订成工艺文件,并安排到相关生产部门。如果在安排生产时发现原辅材料质量存在问题,车间应及时把相关信息反馈给上级部门,以避免危害的进一步扩大化,同时也有利于车间制定防控措施,减小经济损失。
3 结合质量风险管理在无菌药品生产关键步骤
新版GMP 中质量风险评估和管理的理念贯穿了整个无菌药品的生产要求,这概念也逐渐为广大药品生产和质量管理人员所接受。图1为注射剂生产流程及关键点,其将无菌药品生产的整个工艺过程的关键控制点进行了罗列,体现出了整个无菌药品生产质量风险管理的要点。
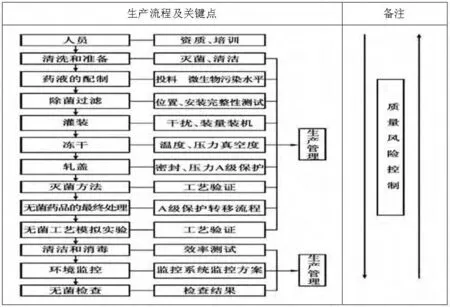
图1 注射剂生产流程及关键点示意图
整个工艺过程关键控制点的质量风险评估方法可用风险评估矩阵,风险发生的可能性(L),后果的严重性(S),可检测性(D),风险等级(R)。具体表达式如下:
风险(R)=风险发生的可能性(L)×后果的严重性(S)×可检测性(D)

表1 风险等级对照及应采取的措施
3.1 质量风险管理在无菌药品生产质量管理中应用实例
3.1.1 药液配制的问题
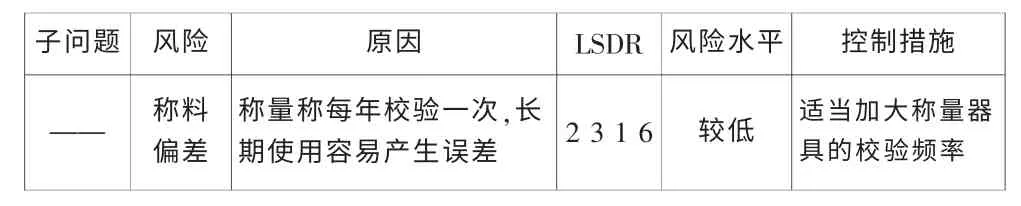
表2
3.1.2 胶塞清洗的问题

表3
3.1.3 冻干工序的问题
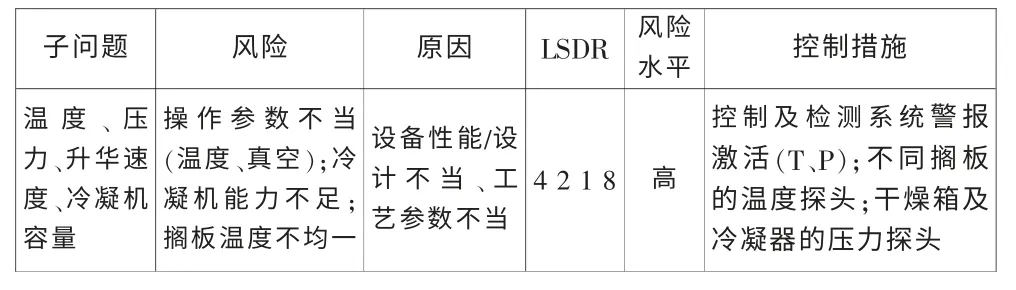
表4
4 加强文件管理和人员培训
在GMP 管理认证过程中,由于时间原因,再加上人手不够,势必会造成管理文件的编写不周全。车间生产是连续的、不断变化的,对于不周全的制度、程序等要及时加以完善并最终以文件的形式保留下来,以适应GMP 管理的发展需求。所以通过认证后更应该严格执行管理文件,执行力度只能比认证前更严格,对于无视制度的员工要做到批评教育,严重者需给予惩处。车间定期或不定期的组织相关人员对生产一线进行检查,检查的内容主要有:生产区域的卫生维护情况;生产过程是否有防尘措施;操作间、容器、设备、物料品种要有明显的状态标识;现场是否有明显的岗位操作规程等。所以,车间的所有员工都要全面系统的进行GMP 培训,务必使每个人都能了解生产全过程,认识和理解自己岗位的角色定位和工作的重要性。对于新招入的员工或新转岗人员要进行专门的培训并考核,不达标的必须进行重新培训直到达标为止,否则不准参加相关的工作。对于个别认识不深刻、工作出现问题较多或以往发生过事故的人员,进行针对性的培训。培训过程可以结合案例进行实际问题剖析,同时要总结出今后正确的工作方法。
5 总结与建议
车间应当对药品生产运行中可能出现的风险因素进行系统的识别,筛选出需要控制的关键影响因素,并根据导致风险的根本原因制定预防措施和监控措施。质量监督员应当明确需要监督的具体风险点、可接受标准监控方式和频率,以此确定监督员的职责,亦可建立现场监督员的报告制度。
[1]国家食品药品监督管理局药品认证管理中心.药品GMP 指南:无菌药品[M].北京:中国医药科技出版社,2011,8.
[2]李秋涛.论药品生产质量管理规范认证后的药品生产管理[J].食品药品监督,2010.
[3]孙悦平.药品生产的现场管理[C]//新版gmp 生产过程控制及现场管理务实培训班.2012,11.
[4]梅占军.2013 年中国药学会药事管理专业委员会年会论文集[C].中国药学会,2013.
