赣西油茶人工林土壤微生物群落的多样性
2015-12-21郭春兰叶素琼吴南生
郭春兰,张 露,叶素琼 ,雷 蕾,吴南生
(1.江西农业大学 园林与艺术学院,江西 南昌 330045;2.江西环境工程职业学院,江西 赣州 341000)
赣西油茶人工林土壤微生物群落的多样性
郭春兰1,张 露1,叶素琼2,雷 蕾1,吴南生1
(1.江西农业大学 园林与艺术学院,江西 南昌 330045;2.江西环境工程职业学院,江西 赣州 341000)
为了解油茶林土壤微生物群落结构特征,揭示油茶林土壤微生物群落结构多样性变化特征对林龄和季节等环境因素的响应机制,为油茶林合理经营管理提供理论依据,以赣西地区新余市渝水区不同林龄油茶林24个土壤样品为研究对象,采用变性梯度凝胶电泳(PCR-DGGE)技术分析油茶林土壤细菌群落多样性。DGGE指纹图谱分析结果表明,油茶林土壤微生物类群的细菌多样性较丰富,各个样品的条带数目稳定分布在13~36条。10年龄油茶人工林细菌基因多样性较为丰富,集约经营10年龄油茶林的细菌物种数量多。细菌丰富度和Shannon-Wiener多样性指数在林龄变化上均表现为中龄林(2个10年龄)>成熟林(30年龄、50年龄)>幼龄林(1年龄、6年龄);其在季节变化上均呈一致的变化规律,为夏季>秋、冬季>春季。细菌16S rDNA的系统发育分析结果表明,大多数序列与未培养细菌的同源性较高,所有序列与数据库中16S rDNA序列的相似性在84%~99%。遗传关系研究得到主要的优势菌为泛菌属Pantoea sp.、肠杆菌属Enterobacter sp.。
油茶林;土壤微生物;PCR-DGGE;细菌群落多样性
土壤微生物是土壤的重要组成部分,不仅能够改善土壤的结构,促进土壤养分转化与循环,更能为地上植被储备养分,促进植物的吸收,增强植物的抗性[1]。微生物群落的组成和活性影响着生物地球化学循环、有机物的代谢过程以及土壤的肥力和质量[2],也制约着林分类型的分异和演替[3]。由于土壤中只有不到l%的微生物是可培养的,且传统培养微生物的方法,很难真实全面地反映土壤微生物群落的变化[4],变性梯度凝胶电泳(PCR-DGGE)技术可以克服传统培养法造成的土壤微生物信息大量丢失的缺点,获得未培养的全部微生物资源,丰富土壤宏基因组文库,并能更精确地揭示土壤微生物种类和遗传多样性[5-7]。近年来,PCR-DGGE分子生物技术广泛应用于微生物多样性研究,涉及湖泊、草地、湿地、熔岩、农田和食品等各个领域,在森林土壤微生物多样性研究中的应用也日趋增多[8-10],这些研究均表明PCR-DGGE技术可以很好地弥补传统培养法的不足,能够较全面分析土壤微生物多样性。目前,油茶林土壤研究主要集中于施肥[11]、套种模式[12]和抚育方式[13]等对油茶林土壤养分及微生物的影响,这些研究主要采用的是传统的化学分析法,且大多数研究仅仅是对油茶林土壤基本养分的探讨[14-16],关于油茶林土壤微生物群落结构多样性的相关报道甚少[17]。因此,本研究中以赣西地区江西省新余市渝水区不同林龄油茶林土壤为研究对象,基于16S rDNA V3区片段的PCR-DGGE技术和克隆测序比对来研究油茶林土壤细菌群落结构特征变化,分析林龄和季节变化对土壤微生物群落结构多样性的影响,以揭示其多样性对林龄和季节的响应机制,为油茶林科学种植管理提供理论依据。
1 材料与方法
1.1 样品采集与研究样地
样品采集标准地按0~20 cm土壤层次,用直径为7.5 cm的土钻在每个样方分别以“S”形采集5个土壤样品,将每个样方的5个样品混合为1个样品带回实验室分析。采集时间分别在2011年10月(秋季),2012年1月(冬季),2012年4月(春季),2012年7月(夏季)。供试材料为6种不同类型油茶人工林及对照灌丛土壤。不同类型油茶人工林主要有幼龄林(1年龄、6年龄),中龄林(2种不同抚育方式的10年龄),成熟林(30年龄、50年龄)。不同抚育方式为集约经营管理和粗放经营管理。
1.2 研究方法
1.2.1 土壤微生物总DNA的提取及检测
方法参照Zhou[18]提出的方法并加以改进,采用化学裂解法直接从土壤样品中提取基因组DNA。
(1)取2 g土壤置于10 mL离心管中,加入裂解液,12 000 r/min离心10 min,弃上清。
(2)向沉淀的土壤中加入3 mL SDS裂解液,200 μL Tris饱和酚和0.3~0.4 g的玻璃珠,涡旋震荡5 min,68 ℃温浴l h,每10 min轻轻摇动1次,12 000 r/min离心10 min。
(3)移取上清液至干净的10 mL离心管中,加入250 μL NH4Ac,-20 ℃至少沉淀15 min,12 000 r/min离心10 min。
(4)移取上清液至干净的10 mL离心管中,加入5 mL苯酚、氯仿、异戊醇混合液(体积比25∶24∶1)和3 mL Tris饱和酚,12 000 r/min离心10 min,重复2次。
(5)移取上清液至1个新的10 mL离心管中,加入等体积异戊醇,-20 ℃放置3 h。
(6)4 ℃下,12 000 r/min离心10 min,弃上清,使用体积分数为70%的冰乙醇,洗涤沉淀2次,自然干燥。加40 μL的TE或无菌水溶解保存备用。
(7)提取的DNA通过1.0%的琼脂糖凝胶电泳检测其DNA完整性。
1.2.2 聚合酶链式反应(PCR)
根据文献[19]~[21],自行设计其特异性扩增引物,序列如表1所示,全部引物均由上海英骏生物技术有限公司合成。PCR反应体系如表2所示。PCR扩增程序为:95 ℃,5 min;94 ℃,30 s;56 ℃,30 s;72 ℃,1 min,共 30 个循环;72 ℃,7 min。
1.2.3 变性梯度凝胶电泳(DGGE)
参照Muyzer等[21]的方法,对16S rDNA的扩增产物进行变性梯度凝胶电泳(DGGE)。使用8%聚丙烯酰胺凝胶,将100%变性梯度定义为包含体积比40%的甲酰胺和7 mol/L尿素。电泳采用DCodeTM Universal Mutation Detection System(Bio-Rad Laboratories,Hercules,CA,USA),首先在220 V电压下预电泳10 min,随后在85 V的固定电压下电泳10 h。电泳结束后,进行硝酸银染色[22]。应用凝胶分析软件Quantity One 4.40对扫描所得的DGGE图谱进行条带识别和分析。
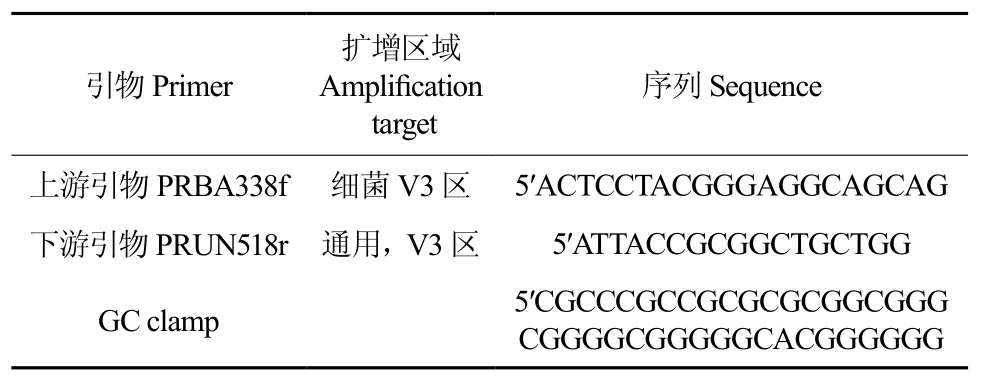
表 1 用于PCR扩增的引物Table 1 Primer sequences for PCR amplification
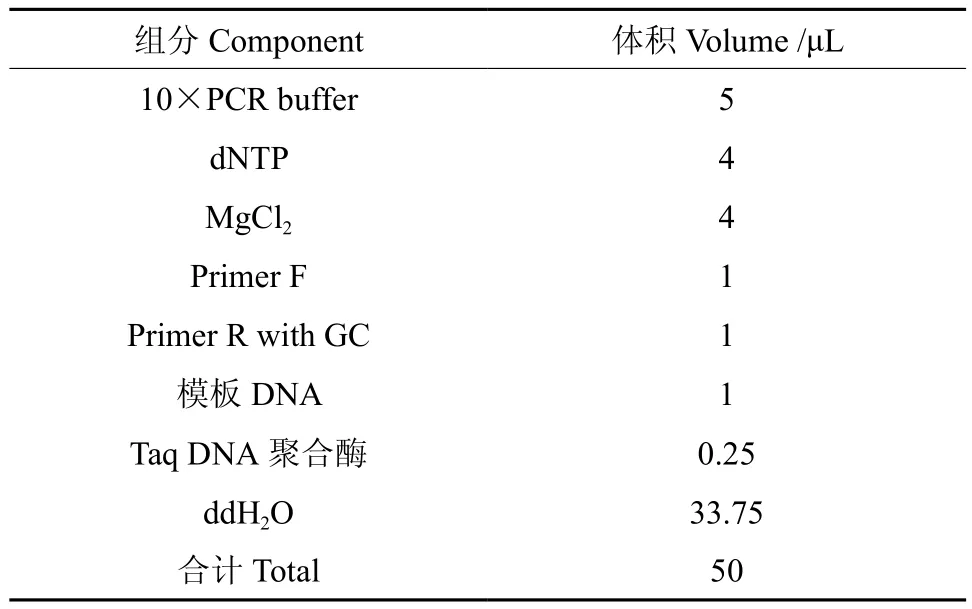
表 2 PCR反应体系Table 2 Reaction system of PCR
1.2.4 条带测序
DGGE特殊条带的测序和分析[23-24]:使用无菌刀片仔细将PCR-DGGE的特定条带从胶上切割下来,使用1×PCR buffer冲洗2次,最后将其捣碎,浸入40 μL 1×PCR buffer中,4 ℃过夜。以其作为PCR模板,重新进行扩增,PCR体系模板量为5 μL,其它条件同1.2.2。取5 μL PCR产物进行1.2%琼脂糖凝胶电泳,阳性结果取10 μL用于DGGE,凡经过重新PCR后的产物在DGGE胶上为单一条带的,然后切胶回收,将PCR产物送生工生物工程(上海)股份有限公司测序部测序。
1.2.5 数据分析
采用Bio-Rad Quantity One 4.40对扫描所得的DGGE图谱进行条带识别和分析,将基因序列均提交 Genbank数据库(http://blast.ncbi.nlm.nih.gov)进行基因序列比对,采用DNAStar,MEGA构建系统发育树。
同时微生物多样性指数的计算公式如下:
(1)香农多样性指数(Shannon's diversity index)

其中,“S”表示总的物种数,“pi”表示第i个种占总数的比例。当群落中只有1个居群存在时,香农指数达最小值0;当群落中有2个以上的居群存在,且每个居群仅有1个成员时,香农指数达到最大值lnk。
(2)物种均一度(Species Evenness)
物种均一度用来描述物种中个体的相对丰富度或所占比例。群落的均一度可以用Pielou均一度指数J表示(Pielou’s evenness index,J):
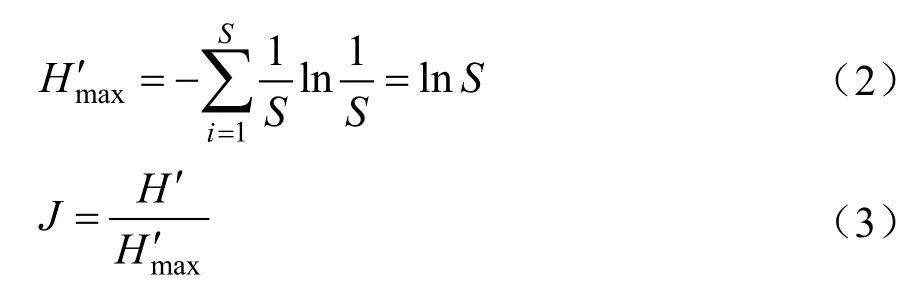
其中,H′为香农指数,H′max是H′的最大值。
2 结果与分析
2.1 细菌总DNA的提取与纯化
从油茶人工林土壤微生物DNA提取的结果来看(见图1),均能提取到23 kb左右的条带,各林分提取的基因组DNA条带明亮,无弥散现象。条带较明亮的林分,表明已获得较长片段的土壤基因组DNA,适合土壤细菌16S rDNA分析。所提取的土壤总DNA中都有部分色素和腐植酸成分,直接影响PCR扩增,进一步对提取的土壤总DNA进行纯化,从而得到质量较高的土壤DNA模板。
2.2 细菌的PCR扩增分析
所提取的DNA溶液纯化后直接用于PCR扩增反应,选择16S rDNA片段带GC夹的338~518作为PCR扩增的引物是进行引物筛选后的结果。DNA溶液纯化并稀释100倍后进行PCR扩增出现了相应的条带,均获得特异性扩增片段,大小在148~169 bp的为16S rDNA V3片段(见图2),能够进行下一步的DGGE图谱分析。
2.3 细菌16S rDNA的DGGE图谱分析
将土壤样品的16S rDNA V3区PCR扩增产物进行DGGE电泳,得到6种类型油茶人工林土壤细菌的DGGE图谱(见图3和图4)。图谱显示,油茶人工林土壤细菌DGGE图谱的条带数和条带位置存在一定程度的差异,土壤样本检出了数目不等、位置各异的电泳条带,说明不同区域相对稳定的微生物群落结构均有差异。电泳图谱中存在共同的电泳谱带,说明这些土壤之间可能存在一些共有的细菌类型。这些条带的分布特征可以说明土壤中既有共同的微生物种类,也有自己独特的微生物种属,形成了各自特定生态位的群落结构[25]。一般来说,DGGE图谱中的每一条带就代表一种微生物物种,优势微生物的条带会明显深一些。图中显示不同类型油茶人工林土壤细菌特征PCR-DGGE图谱,各个泳道中条带数量和亮度均存在差异。从指纹图谱中可以看出,某些条带为不同类型油茶人工林土壤所特有;供试土壤共有的条带也会随着油茶人工林种植年限的不同而呈现出渐弱或渐强。这说明在不同类型的油茶人工林土壤中,土壤细菌群落结构组成发生了改变。
图 1 油茶人工林土壤基因组DNA提取Fig. 1 Soil microbial DNA extraction of different C. oleifera forests
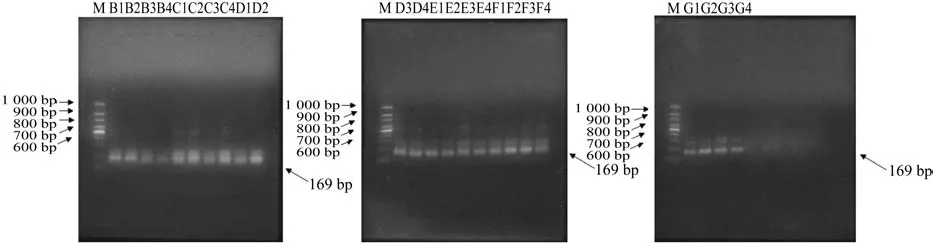
图 2 油茶人工林土壤微生物PCR扩增Fig. 2 Soil microbial PCR ampli fi cation of arti fi cial C. oleifera forests
根据DGGE图谱分析(见图4)后所得的条带数进行Shannon多样性指数分析。由图4可知,不同种植年限、季节变化、管理方式下油茶人工林土壤细菌基因多样性变化特征。从不同种植年限的比较来看,集约经营的1年龄、6年龄和10年龄油茶林土壤的细菌丰富度、香农多样性指数呈现随着种植年限的增长先减少后增加的变化趋势;而粗放经营的10年龄、30年龄和50年龄油茶林则随着种植年限的增长土壤的细菌丰富度、香农多样性指数呈现先增加后减少的变化趋势。
从丰富度和香农多样性指数的季节变化来看,中龄林(D-10yr、E-10yr)和幼龄林(B-1yr、C-6yr)均呈现夏季高,冬季次之,秋、春季较低的变化趋势,成熟林(F-30yr、G-50yr)呈现夏季高,秋、冬季次之,春季较低的变化趋势。
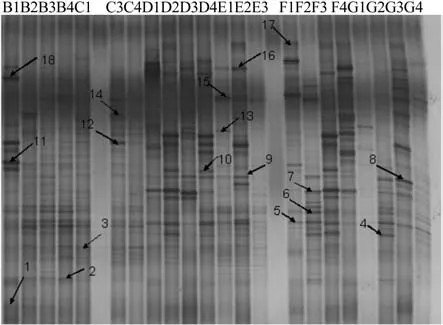
图 3 油茶人工林土壤细菌16S rDNA的DGGE图谱Fig. 3 DGGE spectrum of soil bacterial 16S rDNA of arti fi cial C. oleifera forests
从2种经营管理方式的10年龄油茶人工林(D-10yr,E-10yr)对比来看,集约经营的10年龄油茶人工林(D-10yr)丰富度、香农多样性指数春、夏季均显著大于粗放经营10年龄油茶人工林(E-10yr);且春、夏季丰富度均值依次提高8.33%,16.67%;春、夏季香农多样性指数均值依次提高2.94%,5.11%;秋季差异不显著。
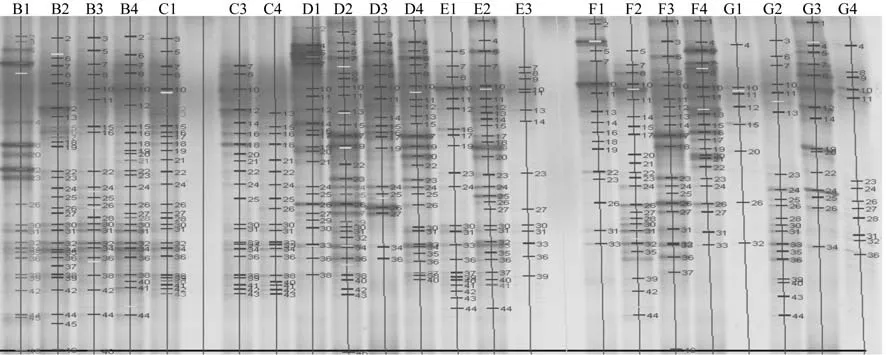
图 4 油茶人工林土壤细菌16S rDNA的DGGE图谱泳道分析Fig. 4 Lane analysis of DGGE spectrum of soil bacterial 16S rDNA of arti fi cial C. oleifera forests
2.4 细菌16S rDNA片段的DGGE指纹图谱的聚类分析
根据各条带的相对迁移率和相对密度进行聚类分析(UPGMA),各供试土壤之间相似性系数的聚类分析结果见图5。在6种类型的油茶人工林土壤中,细菌群落组成表现出较大的变异性,其各自的优势条带明显,相似性为25%~70%,同时表现出明显的季节变化,说明油茶人工林从幼龄林、中龄林至成熟林群落结构发生了显著变化。
2.5 细菌16S rDNA的系统发育分析
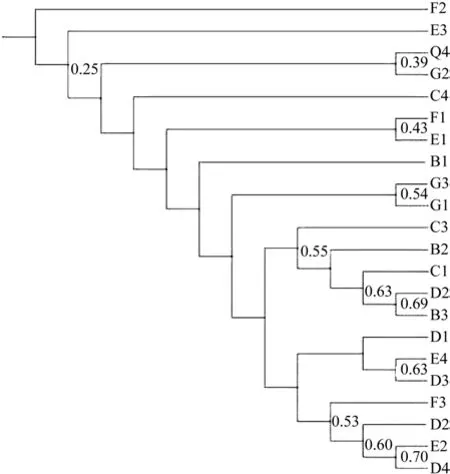
图 5 油茶人工林土壤细菌群落多样性指数的聚类分析Fig. 5 Cluster analysis of soil bacteria community diversity index of arti fi cial C. oleifera forests
细菌16S rDNA已成为目前细菌系统分类研究中最常用的分子指标,测定16S rDNA部分序列可达到对分离细菌分子鉴定的目的[26-27]。本研究中将DGGE图谱上的优势条带切割下来进行纯化克隆测序,将分离菌株的16S rDNA序列与GenBank数据库中核酸序列进行相似性比对,结果见表3。由表3可知,大多数序列与未培养细菌的同源性较高,所有序列与数据库中16S rDNA序列的相似性在84%~99%。其中,肠杆菌属Enterobacter ludwigii与泛菌属Pantoeasp、未培养芽孢杆菌属Uncultured Bacillussp等菌株的16S rDNA序列相似性高达97%~99%,优势条带所对应的序列登录号见表3。
将所得序列及其相似序列进行遗传关系研究,采用邻接法构建系统发育树状图(见图6),得到优势菌属为泛菌属Pantoeasp、肠杆菌Enterobacter ludwigii、菠萝泛菌Pantoea ananatis。
3 讨论与小结
(1)本研究结果是随着种植年限的增长,油茶人工林土壤微生物群落基因多样性指数的集约经营的1年龄、6年龄和10年龄呈现为随着种植年限的增长先减少后增加的变化趋势,而粗放经营的10年龄、30年龄和50年龄则随着种植年限的增长呈现先增加后减少的变化趋势。刘丽等[28]在研究不同发育阶段杉木人工林对土壤微生物群落结构的影响中得出,杉木人工林土壤微生物群落多样性指数随着杉木人工林的生长发育而逐渐增加,这与本研究结果不完全一致。王雪山等[29]在研究种植年限对牡丹根际土壤微生物群落结构的影响中得出,土壤微生物群落结构多样性水平随种植年限的增加而降低,菌群结构趋于简单,种类大量减少。其原因主要是随着种植年限的增长,树体每一生长发育阶段的特性改变了土壤微域生境条件,为微生物的生长提供了丰富的碳源和氮源,引起土壤微生物群落结构的变化。
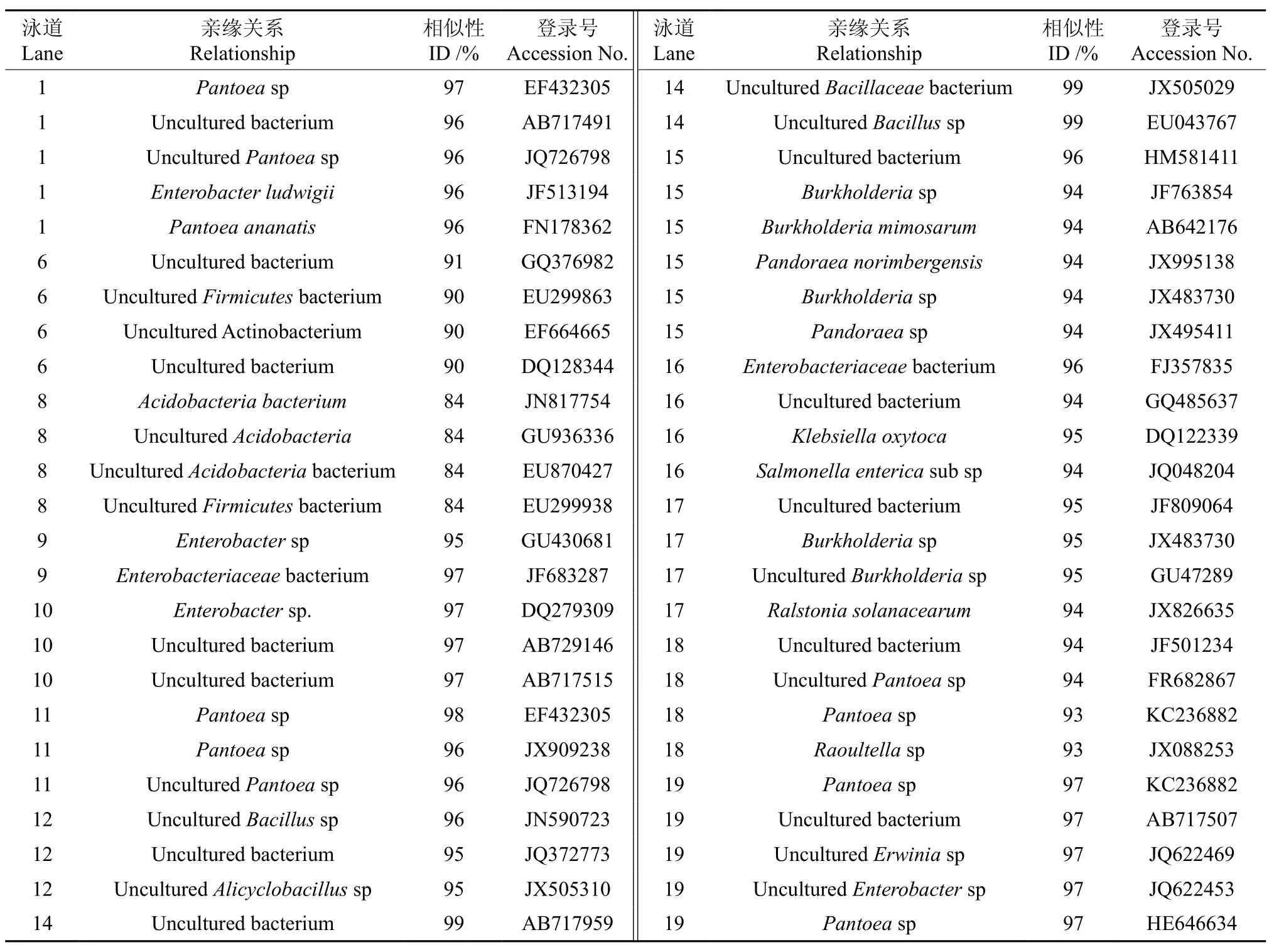
表 3 核苷酸序列同源性比对†Table 3 Homology comparison of nucleotide sequences
(2)本研究中,中龄林与成熟林、幼龄林呈现夏季高,春季较低的变化趋势,这一结果和前人的研究结果[30-31]相似。Schmidt等[32]的研究表明,在1年之内土壤微生物群落演替的主要原因是土壤微生物利用的主要底物发生了季节性演替:碳聚合物或酚类(冬季)、蛋白质(春季)、根际沉积物(夏季);这也说明在不同的森林生态系统中,由于各种生态因子复杂的综合作用以及关键生态因子的主导地位的不同,土壤微生物群落季节动态变化存在着显著差异。Petra Marschnera等[33]认为,土壤有机碳的含量和C/N比能够显著影响土壤细菌群落结构的变化。由此可知,季节变化影响土壤的微域生境,从而导致不同类型的土壤微生物群落结构的相似性或多样性变化。
(3)土壤微生物多样性的影响因素很多,如土壤营养状况、质地、温度、水分和通气性等,一定的集约经营管理通过影响土壤含水量、温度、通气性、pH值及有机碳和氮的水平而影响土壤微生物多样性。本研究中集约经营10年龄油茶人工林丰富度、香农多样性指数在春季和夏季均显著大于粗放经营10年龄油茶人工林,且依次提高12.50%和4.03%。集约经营10年龄油茶人工林的菌种数量多,且在油茶人工林土壤中的优势地位较明显。刘琛等[34]在围垦海涂微生物群落和土壤酶特征研究中得出,围垦利用后土壤质量优于未利用海涂,土壤微生物多样性指数高,该结果与本研究有相似之处。这主要是因为一定的集约经营管理方式改变了土壤微生境条件,引起土壤微生物群落结构的变化,对土壤的微生物生态环境产生了影响,同时不同程度地提高群落中各物种分布的均匀度与异质性。集约经营管理改变了林内的光照、温度等生境条件,减少了植物种内对养分和水分的竞争,增加了物种丰富度,改善了土壤质量,有利于改善微生物的繁育条件。由此可知,集约经营管理措施通过改善土壤质量,能提高油茶人工林土壤微生物丰富度,增加微生物活性。
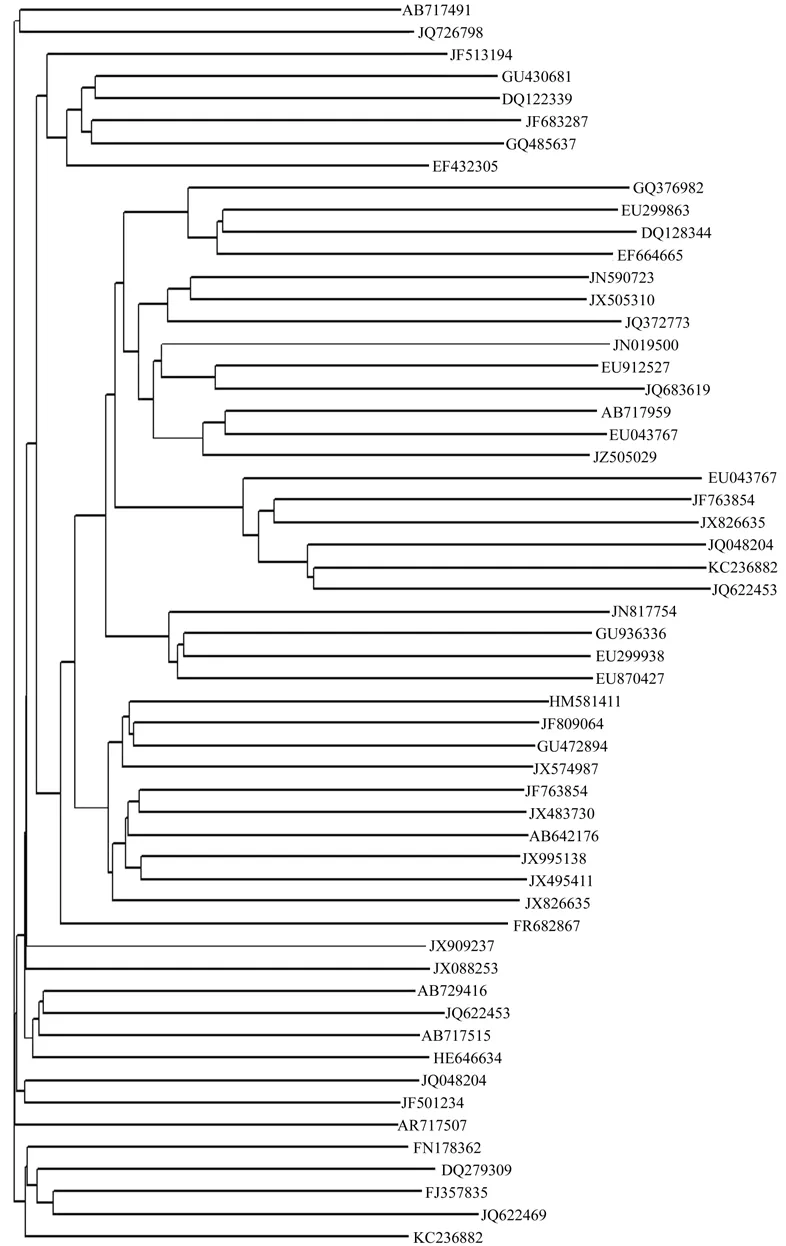
图 6 系统发育树Fig. 6 Phylogenetic tree
(4)PCR-DGGE技术是一种免培养的方法,从土壤微生物基因组的角度研究其多样性。有必要进一步从遗传和基因表达水平上对土壤微生物生理生化特性、微生物群落结构及多样性等相互作用进行研究,对于微生物哪些种属在影响土壤肥力时起了主要作用,是否可以分离筛选出有益微生物并用于生产实践,有必要加强以功能基因组为核心的土壤微生物分子生态研究。本研究中只对细菌基因多样性进行了探索,下一步将对其它重要独特生理特群(如真菌、放线菌以及古菌等)进行试验,通过克隆测序鉴定出各自具体的种属,以诊断和评价复杂微生物群落的种群结构。优势条带测序结果与GenBank数据库序列进行Blast序列比对,可以确定油茶人工林土壤中的优势菌属。所有序列与16S rDNA序列的相似性在84%~99%,其中与泛菌属Pantoeasp、肠杆菌属Enterobactersp、未培养芽孢杆菌属Uncultured Bacillussp等菌株的16S rDNA序列相似性高达97%~99%。
[1]林先贵,胡君利.土壤微生物多样性的科学内涵及其生态服务功能[J].土壤学报,2008,45(5):892-900.
[2]刘权钢,金东淳,刘敬爱.DGGE技术在土壤微生物多样性分析上的研究进展[J].延边大学农学学报,2012,34(2):170-177.
[3]刘绍雄,王明月,王 娟,等.基于PCR-DGGE技术的剑湖湿地湖滨带土壤微生物群落结构多样性分析[J].农业环境科学学报 ,2013,32(7):1405 - 1412.
[4]Vres L, Torsvik V. Microbial diversity and community structure in two different agricultural soil communities[J]. Microb Ecol,1998, 36: 303 - 315.
[5]Chong C W, Tan G A, Wong R C,et al.DGGE fi ngerprinting of bacteria in soils from eight ecologically different sites around Casey Station, Antarctica[J]. Polar biology, 2009, 32(6): 853 - 860.
[6]滕 应,骆永明,赵祥伟,等.重金属复合污染农田土壤DNA的快速提取及其PCR-DGGE分析[J].土壤学报,2004,41(3):343-347.
[7]李 丹,王秋玉.变性梯度凝胶电泳及其在土壤微生物生态学中的应用[J].中国农学通报,2011,27(3): 6-9.
[8]Kornelia S, Miruna OS, Annett M,et al.Bacterial diversity of soil assessed by DGGE, T-RFLP and SSCP fi ngerprints of PCR-ampli fi ed 16S rRNA gene fragments: Do the different methods provide similar results [J]. Journal of Microbiological Methods,2007, 69(3): 470 - 479.
[9]Xue D, Yao H Y, Ge DY,et al.Structural and functional diversity of bacterial community in soil treated with the herbicide napropamide estimated by the DGGE, CLPP and r/K-strategy approaches[J]. Applied Soil Ecology, 2013, 72: 242 - 250.
[10]Zhu L G, Xu H T, Zhang Y P,et al. BOX-PCR and PCR-DGGE analysis for bacterial diversity of a naturally fermented functional food (Enzyme®)[J]. Food Bioscience, 2014, 5:115 - 122.
[11]何 钢,袁德义,刘贤桂.油茶低产林土壤改良对土壤养分及土壤酶活的影响[J].中南林业科技大学学报,2011,31(3):76-80.
[12]李纪元,肖 青,李辛雷,等.不同套种模式油茶幼林水土流失及养分损耗[J].林业科学,2008,44(4):167-172.
[13]郝 艳,刘君昂,周国英,等.不同抚育方式对油茶人工林土壤养分、微生物及酶活性的影响[J].林业资源管理, 2008,(6): 97 - 101.
[14]周招娣,张日清,马锦林,等.6个油茶物种苗期抗旱性的初步研究[J].经济林研究,2014,32(2):53—57.
[15]滕维超,刘少轩,曹福亮,等.油茶大豆间作对盆栽土壤化学和生物性质的影响[J].中南林业科技大学学报, 2013, 33(2):24—27.
[16]魏世清,伍 琪,黄凌志,等.沼肥配施复合肥对油茶生长及产量的影响[J].中南林业科技大学学报,2014,34(3):53—57.
[17]郭春兰,张 露,雷 蕾,等.不同林龄油茶人工林土壤酶活性及养分特征[J].草业科学,2012,29(11):1647-1654.
[18]Zhou J, Bruns M A, Tiedje J M,et al.DNA recovery from soils of diverse composition[J]. Appl Environ Microbial, 1996, 62(2):316-322.
[19]Jeyaram K, Mohendro Singh W, Premarani T,et al. Molecular identi fi cation of dominant micro fl ora associated with Hawaijar′-A traditional fermented soybean (Glycine maxL) food of Manipur,India [J]. Int J Food Microbiol,2008,122(3):259 - 268.
[20]Nübel U, Engelen B, Felske A,et al.Sequence heterogeneities of genes encoding 16S rRNAs inPaenibacillus polymyxadetected by temperature gradient gel electrophoresis [J]. J Bacteriol, 1996,178(19):5636 - 5643.
[21]Muyzer G, Dewaal E C, Uitterlinden A G. Pro fi ling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-ampli fi ed genes coding for 16S rRNA[J]. Appl Environ Microbiol, 1993, 59(3):695 - 700.
[22]汪孟娟,熊顺强,陈廷涛,等.PCR-DGGE监测豆豉制曲过程中菌群的动态变化[J].南昌大学学报:理科版, 2010, 34(6):571-574,578.
[23]Devillard E, Burton J, Reid G. Complexity of vaginal micro fl ora as analyzed by PCR denaturing gradient gel electrophoresis in a patient with recurrent bacterial vaginosis [J]. Infectious Diseases in Obstetrics and Gynecology, 2005, 13(1):25 - 30.
[24]Nübel U, Engelen B, Felske A,et al.Sequence heterogeneities of genes encoding 16S rRNAs inPaenibacillus polymyxadetected by temperature gradient gel electrophoresis [J]. J Bacteriol, 1996,178(19):5636 - 5643.
[25]刘新宇.土壤细菌16S rRNA基因V3区种属特异性检测的法医学应用研究[D].沈阳:中国医科大学,2008.
[26]戴 欣,陈月琴,周 惠,等.海洋细菌的分子鉴定分类[J].中山大学学报:自然科学版,2000,39(1):68-71.
[27]Anthony G O Donnell, Heike E Gorres. 16S rDNA methods in soil microbiology [J]. Current Opinion in Biotechnology, 1999,10: 225 - 229.
[28]刘 丽,段争虎,汪思龙,等.不同发育阶段杉木人工林对土壤微生物群落结构的影响[J].生态学杂志, 2009, 28(12):2417-2423.
[29]王雪山,杜秉海,姚良同,等.种植年限对牡丹根际土壤微生物群落结构的影响[J].山东农业大学学报:自然科学版,2012, 43(4):508 - 516.
[30]Schmidt S K, Lipson D A. Microbial growth under the snow:Implications for nutrient and alleochemical availability in temperate soils[J]. Plant and Soil, 2004,259:1 - 7.
[31]Wallenstein M D, Mcmahon S, Schinel J. Bacterial and fungal community structure in Arctic tundra tussock and shrub soils [J].FEMS Microbiology Ecology, 2007,59(2):428 - 435.
[32]Schmidt S K, Costello E K, Nemergut D R,et al.Biogeochemical consequences of rapid microbial diversity: A review[J].Biodiversity Science, 2004, 12(4):456 - 465.
[33]Petra Marschnera, Ellen Kandeler, Bernd Marschner. Structure and function of the soil microbial community in a long-term fertilizer experiment[J]. Soil Biology and Biochemistry, 2003,35(3): 453 - 461.
[34]刘 琛,丁能飞,郭 彬,等.不同土地利用方式下围垦海涂微生物群落和土壤酶特征[J].土壤通报,2013,44(1):99-106.
Soil microbial community diversity of Camellia oleifera forest in western Jiangxi
GUO Chun-lan1, ZHANG Lu1, YE Su-qiong2, LEI Lei1, WU Nan-sheng1
(1.College of Landscape and Art, Jiangxi Agricultural University, Nanchang 330045, Jiangxi, China;2.Jiangxi Environmental Engineering Vocational College, Ganzhou 341000, Jiangxi, China)
In order to learn the characteristics of soil microbial community structure of Camellia oleifera forest, and to reveal the responding mechanism of variation characteristics of soil microbial community structure diversity of C.oleifera forest on the environmental factors of forest ages, seasons, and so on, and to provide a theoretical basis for reasonable operation and management of C. oleifera forest, taking 24 soil samples in C. oleifera forest at different ages at Yushui District of Xinyu City of western Jiangxi as research objects, soil bacterial community diversity of C. oleifera forest was analyzed by using PCR-DGGE technology. The results of DGGE fi ngerprint analysis showed that soil bacterial diversity of C. oleifera forest was relatively rich, and the number of bands of each sample was from 13 to 36. The soil bacterial genetic diversity of ten-year arti fi cial forest was the most abundant, and number of bacterial strains in ten-year forest through intensive operation was the most. Based on bacterial richness and Shannon-Wiener diversity index from high to low, the order was middle age forest (two 10-year forests), mature forest (30-year and 50-year forests), and young forest (1-year and 6-year forests). The two indexes showed a seasonal variation consistent, the values in summer were higher than those in autumn and winter, and the values were the lowest in spring. The results of phylogenetic analysis of bacterial 16S rDNA showed that, most sequences had high homology with uncultured bacteria, and the similarity of the 16S rDNA sequences was between 84%-99%. The results of genetic relationship analysis showed that, the dominant bacteria werePantoeasp andEnterobactersp.
Camellia oleiferaforest; soil microorganism; PCR-DGGE; bacterial community diversity
S606+.1;S794.4 文献标志码:A 文章编号:1003—8981(2015)01—0025—08
2014-10-03
江西省青年科学基金项目(20122BAB213018);江西省研究生创新基金(YC2011-S058);江西农业大学博士启动基金;国家科技部“十一五”科技支撑项目(2009BADB1B05-03);江西省科技创新“六个一”工程重大科技专项“江西省油茶产业升级关键技术研究与示范”(赣科发计字〔2009〕230号)。
郭春兰,讲师,博士。
吴南生,副教授。E-mail:362220279@qq.com
郭春兰,张 露,叶素琼,等.赣西油茶人工林土壤微生物群落多样性研究[J].经济林研究,2015,33(1):25-32.
[本文编校:闻 丽]
