静电纺丝法制备交联多孔纳米碳纤维膜及其电化学电容性能
2015-12-07卢建建应宗荣刘信东赵双生
卢建建 应宗荣 刘信东 赵双生
(南京理工大学化工学院, 南京 210094)
静电纺丝法制备交联多孔纳米碳纤维膜及其电化学电容性能
卢建建 应宗荣*刘信东 赵双生
(南京理工大学化工学院, 南京 210094)
以聚丙烯腈(PAN)和三聚氰胺为原料, 通过静电纺丝法制备了三聚氰胺改性聚丙烯腈纳米纤维前驱体,经预氧化、碳化后得到交联的多孔纳米碳纤维. 采用红外光谱(FTIR)仪、热重分析(TGA)仪、扫描电子显微镜(SEM)、X射线衍射(XRD)仪、拉曼光谱仪和比表面积分析仪等对前驱体及纤维进行了表征. 结果表明, 经过三聚氰胺改性的聚丙烯腈纳米纤维前驱体在碳化后有效地交联, 形成含有微孔、介孔和大孔多级的合理孔道结构, 氮掺杂量高达14.3%, 纤维直径大幅缩减, 平均直径仅约89 nm. 电化学测试结果表明, 交联多孔纳米碳纤维电极在0.05 Ag–1电流密度下未经活化时的质量比电容值高达194 Fg–1(0.05 Ag–1), 在2 Ag–1的电流密度下经过1000次循环充放电后的比电容仍然保持99.2%, 表现出优异的电化学特性.
三聚氰胺; 静电纺丝; 纳米碳纤维; 交联网络; 超级电容器
1 引 言
电化学电容器也叫超级电容器, 是一种介于传统电容器和电池之间的新型储能器件, 因其具有高功率密度、长循环寿命、快速充放电速率、低的制造成本等优点而引起广泛关注.1–3按照储能机理的不同, 超级电容器可以分为两类: 双电层电容器和法拉第赝电容器.4电极材料是决定电容器比电容大小的关键因素, 赝电容电容器虽然表现出很高的比电容, 然而存在循环稳定性差、电导率低、价格昂贵等问题,5限制了其大规模的应用. 双电层电容器电极材料主要是各种碳材料, 其中包括活性炭、6碳纳米管、石墨烯7,8和多孔纳米碳纤维9等. 大量的研究表明, 大的比表面积、高电导率、合理的孔道分布以及杂原子的掺杂对提高碳材料的双电层电容性能意义重大.10
纳米碳纤维作为准一维纳米碳材料近年来受到广泛关注.11特别是多孔纳米碳纤维因独特的纤维结构, 不仅具有高的孔隙率、大的比表面积和大长径比等优点, 还兼具低密度、高比模量、高比强度、高导电性和高导热性等特性,12因而在储存材料、电极材料、催化剂和催化剂载体、高效吸附剂、分离剂以及复合材料等方面具有广阔的应用前景.13–15静电纺丝法结合碳化工艺制备多孔纳米碳纤维因工艺简单、成本低廉而成为制备该种材料最有效的方法之一.16Jung等17电纺聚丙烯腈溶液, 碳化后得到纳米碳纤维, 分别用氢氧化钾和氢氧化钠在高温惰性气氛下对纳米碳纤维进行活化得到多孔纳米碳纤维, 并将其应用作电容器电极材料. Kim等18将原硅酸四乙酯和聚丙烯腈混溶电纺, 活化后得到含硅的多孔纳米碳纤维, 然而将其用作电化学电容器电极材料时比电容仅有92.0 Fg–1. Park 等19电纺聚丙烯腈和聚苯乙烯的混合溶液制备氮掺杂多孔纳米碳纤维, 耐热性能较差的聚苯乙烯作为成孔剂, 在高温下分解制得高比表面积纳米碳纤维,然而聚丙烯腈本身含氮量较低, 碳化过程中氮的含量还会进一步降低, 因而制备得到的氮掺杂纳米碳纤维的氮含量很低, 以至无法有效地引入赝电容.
鉴于活化过程涉及复杂的化学反应且程序繁琐, 因而不利于多孔碳纤维的制备. 本文采用三聚氰胺作为交联改性剂和氮掺杂剂, 通过共混静电纺丝法制备了三聚氰胺/聚丙烯腈纳米纤维前驱体, 再经碳化工艺一步法制备得到了高氮掺杂纳米碳纤维. 利用三聚氰胺的熔融热分解特性, 采用合适的预氧化工艺, 使纳米纤维前驱体在还未通过充分预氧化使形貌固定的情况下, 借助三聚氰胺的熔融热分解导致纳米纤维塌陷和收缩, 使纳米纤维有效地扁化和粘并, 形成交联三维立体网络状结构, 产生各种尺寸不一的介孔和大孔, 与此同时三聚氰胺发生热分解, 其产生的气体在软化的有机纤维内部形成微孔, 使最终纳米碳纤维内部同时具有大量微孔,两者共同导致纳米碳纤维具有合理的多级孔道结构和较大的比表面积. 特别是, 由于特殊的交联三维立体网络状结构导致网络中纳米纤维搭接面积增加并成为一体, 提供良好的连续导电网络结构,增强电子的传导能力, 最终纳米碳纤维膜的电阻大大降低, 从而提高了其电化学特性. 本文探讨了三聚氰胺对电极电容性能的影响, 发现三聚氰胺氮掺杂纳米碳纤维的质量比电容值高达194 Fg–1(电流密度为0.05 Ag–1), 并且电化学稳定性优良, 表现出优异的电化学电容特性.

图1 纳米碳纤维电极的制备过程示意图Fig.1 Schematic illustration of the synthesis of carbon nanofibers electrode
2 实验部分
2.1 实验原料
三聚氰胺(国药集团化学试剂有限公司, 化学纯);聚丙烯腈(上海金山石化有限公司, 分子量为53000); N,N-二甲基甲酰胺(DMF, 国药集团化学试剂有限公司, 分析纯); 氢氧化钾(南京化学试剂有限公司, 分析纯); 纯氮气(南京文达特种气体有限公司, 99.99%).所有试剂均未经进一步纯化直接使用.
2.2 纳米碳纤维的制备
氮掺杂多孔纳米碳纤维的制备过程如图1所示.将不同质量的三聚氰胺加入60 °C的21.5 g DMF中充分搅拌2 h后, 加入相应质量的聚丙烯腈(PAN)粉末(三聚氰胺和PAN的质量比分别为0 : 1、1 : 8、1 : 4, 溶质总质量为3.5 g), 继续搅拌12 h后, 所得不同质量配比的三聚氰胺乳液作为电纺溶液, 并分别标记为 M1、M2和M3. 静电纺丝条件为:正高压20 kV,针头与接收器之间的距离为15 cm, 流速0.5 mLh–1,纺丝环境温度为室温25 °C左右. 连续静电纺丝3–5 h后, 取下接收器并将纺制的纤维膜放入电热鼓风干燥箱内70 °C干燥12 h备用. 将干燥好的有机纤维膜裁剪成合适尺寸放入瓷方舟中并放置于GSL1400 X真空管式高温烧结炉(合肥科晶材料技术有限公司)内. 首先在大气环境下以3 °Cmin–1的升温速率升温到290 °C, 并保温90 min进行预氧化, 然后将管式炉内通入氮气, 在氮气保护下以3 °Cmin–1的升温速率升温到420 °C, 并保温120 min, 随后升温到800 °C碳化1 h, 最后以5 °Cmin–1的降温速率冷却到室温得到纳米碳纤维. M1、M2和M3电纺溶液得到的纳米碳纤维分别标记为PAN、PM1和PM2纳米碳纤维.
2.3 纳米碳纤维的结构表征
采用FTIR-8400S型傅里叶变换红外分析仪(SHIMADZU)进行红外光谱测试; 热重分析采用DTG-60热重和差热同步仪(SHIMADZU)以10 °Cmin–1升温速率在30–800 °C温度范围内测试; 扫描电子显微镜观察采用S-4800II场发射扫描电子显微镜(Hitachi High-Technologies Corporation, Japan); 采用Elementar Vario MICRO(德国)有机元素分析仪对三聚氰胺改性纳米碳纤维进行氮元素含量的测定; 采用PHI5300(Perkin-Elmer)X射线光电子能谱仪对纳米碳纤维样品进行X射线光电子(XPS)测试, 以波长为457 nm的Al Ka靶(hv = 1486.7 eV)为激发光源; 采用D8 Advance X射线衍射仪(Bruker AXS GmbH, Germany)对纳米碳纤维膜进行测试, 靶材为Cu靶,工作电压及电流分别为40 kV和40 mA, 2θ角度范围为10°–80°; 拉曼光谱测试采用Renishaw (in Via)激光共焦显微拉曼光谱仪(英国雷尼绍公司)进行测试;比表面积测试采用ASAP2020系列全自动物理化学吸附仪(美国麦克)在液氮(77 K)条件下测定样品的氮气吸附-脱附等温曲线及孔径分布曲线.
2.4 纳米碳纤维的电化学测试
按照8:1:1的质量比分别称取纳米碳纤维样品、乙炔黑和聚四氟乙烯(PTFE)乳液, 控制总质量在10 mg左右, 充分研磨后涂在泡沫镍样品上, 60 °C干燥12 h后在10 MPa压强下压制成电极片. 以6 molL–1KOH水溶液为电解液, 铂片电极为对电极,饱和甘汞电极(SCE)为参比电极以及纳米碳纤维为工作电极组成三电极体系, 用Autolab电化学工作站在–0.9―0 V的电位区间内分别进行循环伏安(CV)、恒流充放电(CP)和电化学阻抗(EIS)测试.
3 结果与讨论
3.1 电纺前驱体有机纤维的FTIR及TG分析
将PAN、PM1和PM2电纺前驱体有机纤维膜进行红外光谱分析, 结果如图2所示. 从图2可见, 前驱体有机纤维膜的红外光谱在2241 cm–1处均存在红外吸收峰, 对应于聚丙烯腈中氰基(CN)三键的伸缩振动, 而且随着三聚氰胺比例的增大, 该处的吸收峰依次减小. 而PAN、PM1和PM2电纺前驱体有机纤维膜在3468和3406 cm–1处出现了三聚氰胺的伯胺(NH2)伸缩振动峰, 1620 cm–1处出现了NH2扭曲振动峰, 1523和1431 cm–1处出现了三嗪环伸缩振动的特征吸收峰,20对应于三聚氰胺中的三嗪环, 这说明三聚氰胺被成功添加进入电纺纳米纤维中.

图2 PAN、PM1和PM2电纺前驱体有机纤维的傅里叶变换红外光谱图Fig.2 FTIR spectra of PAN, PM1, and PM2 electrospinning precursor organic nanofibers
PAN前驱体有机纤维的TGA曲线(图3)在30–100 °C的失重是由于表面吸附水的脱附所致, 失重率大约在2%; 在305–480 °C的温度范围内PAN纳米纤维出现剧烈失重, 这主要是由于聚丙烯腈分子链间发生一系列复杂的交联、环化及缩聚反应, 释放出NH3、CO、CO2、H2、N2和H2O等小分子导致了质量损失. 而在480–800 °C 范围内纳米纤维的失重较为平缓, 这主要是纳米纤维的进一步脱N和H元素导致的质量损失.21而PM1和PM2电纺前驱体有机纤维剧烈失重的温度区间为250–300 °C, 这主要是由于三聚氰胺的加入在受热时熔融分解释放出氨气、氰化氢等气体所致.22而在300–800 °C的失重主要是聚丙烯腈分子间复杂的缩聚反应所致.

图3 PAN、PM1和PM2电纺前驱体有机纳米纤维的TGA曲线Fig.3 TGA curves of PAN, PM1, and PM2 electrospinning precursor organic nanofibers

图4 电纺前驱体有机纳米纤维及纳米碳纤维的SEM照片Fig.4 SEM images of electrospinning precursor organic nanofibers and carbon nanofibers
3.2 电纺前驱体有机纤维及其纳米碳纤维的SEM分析
从图4(B, F)可见, PAN电纺前驱体有机纤维的平均直径为256 nm, 碳化后为189 nm, 缩减了大约30%. 从图4(C, G)发现, PM1电纺前驱体有机纤维的平均直径为234 nm, 碳化后为134 nm, 缩减了大约43%. 而PM2电纺前驱体有机纤维的平均直径为166 nm, 碳化后为89 nm, 缩减了大约46%(图4(D, H)). 从TGA的测试结果我们发现, 碳化后三聚氰胺掺杂的纳米纤维的剩余率比纯组分PAN纳米纤维要低, PAN、PM1和PM2电纺前驱体有机纤维800 °C 时的残炭率分别为43.7%、38.7% 和38.3%, 这主要归因于三聚氰胺的分解, 使纤维失重较大, 因此纤维收缩细化明显, 这与TG的测试结果相吻合. 从图4(B, C, D)发现, 随着三聚氰胺含量的增加, 电纺前驱体有机纤维表面粗糙度增加, 这是由于三聚氰胺不溶于DMF溶液中, 而是以微粒悬浮分散于PAN的DMF溶液里, 以致电纺液是浑浊的分散液, 包含于电纺纤维皮层的三聚氰胺微粒导致纤维表面凸起,以致粗糙度随其含量而增加. 这一现象也说明我们成功纺制了含有三聚氰胺的前驱体有机纳米纤维,这一点在红外光谱中也得到确认. 从图4(F, G, H)我们还发现, 三聚氰胺的加入导致碳化后的纳米碳纤维出现扁化, 并且纤维之间发生明显的粘并. 我们推测, 由于我们选择的预氧化温度为290 °C, 而在250 °C时三聚氰胺就已经开始熔融分解, 此时PAN有机纤维还未充分预氧化而固定形貌, 三聚氰胺的熔融分解导致纳米碳纤维收缩和塌陷, 使纳米纤维发生扁化、粘并和交联, 形成三维立体网络状结构, 产生各种尺寸不一的介孔和大孔, 同时由于这样的特殊结构导致网络中纳米纤维搭接面积增加, 降低接触电阻和增大接触通路, 提供良好的连续导电网络结构, 增强电子的传导能力, 最终纳米碳纤维膜的电阻大大降低, 从而提高了其电极电化学特性.23,24
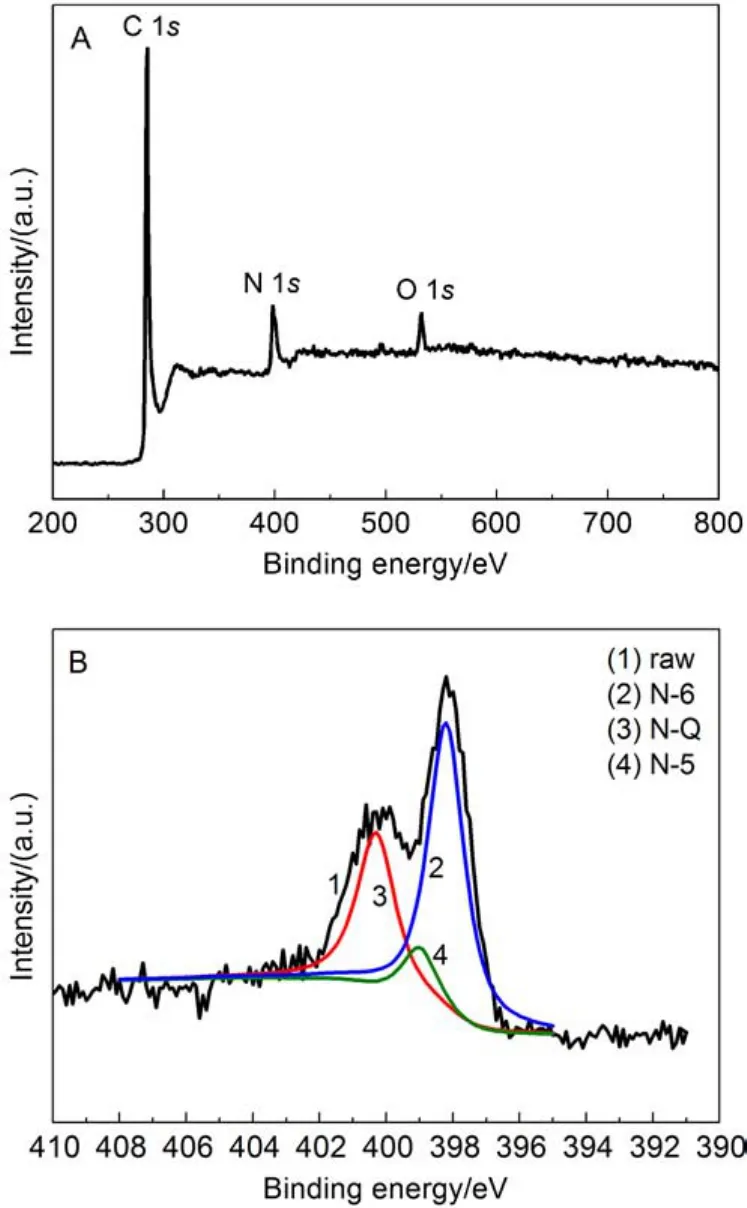
图5 PM2纳米碳纤维的XPS全谱图(A)和N 1s的精细谱图 (B)Fig.5 XPS survey scan spectum (A) and N 1s fine scan spectra (B) of PM2
3.3 纳米碳纤维的N含量测定及XPS分析
为了探讨氮元素的掺杂量对电极比电容的影响, 对三聚氰胺改性纳米碳纤维样品进行了元素分析. 结果表明PM1和PM2纳米碳纤维中的N含量分别为10.6%和14.3%(w). 相关的文献报道制备N掺杂纳米碳材料的含氮量高低不一, 通过NH3在高温下活化纳米碳纤维得到的含氮量仅为3.3%;25通过三聚氰胺与其他试剂协同分解制备的氮掺杂纳米碳材料的含氮量为7.72%.26而未改性聚丙烯腈基纳米碳纤维的N含量一般低于4%.27元素分析结果表明,我们通过三聚氰胺乳状液作为电纺溶液实现了三聚氰胺与聚丙烯腈的高比例混纺, 得到了内部分散有三聚氰胺的微纳米纤维膜, 结合碳化工艺制备的纳米碳纤维可以有效实现高氮掺杂, 高含量的氮掺杂提高了表面润湿性, 提供法拉第赝电容,28使碳材料获得高的电化学特性, 比如高比电容.
为了进一步确定N元素的存在形态, 我们对PM2样品进行了XPS的测试分析. 从图5A中可以看出C、N和O三种元素的信号峰, 并且N元素的含量相对较高, 同时也可以发现一小部分的氧元素的信号峰, 这部分的氧元素有可能是纳米碳纤维表面所吸附的氧气所致.29图5B给出了N1s的精细谱图, 图中结合能在398.2、399.1和400.3eV处可分别归类为吡啶型(N-6)、吡咯型(N-5)和季氮(N-Q)等类型的氮. 根据之前的报道,30,31在这三种类型的氮中, NQ型和N-6型氮均为sp2杂化类型的氮, 这可以提高碳材料的电导率, 并且可以因此增强碳材料的电化学性能. 此外, 由于N-6型氮原子可以引起大量的缺陷,这些缺陷可以增加电荷的存储能力. 由图中N1s元素的精细谱图可知, N-6、N-5和N-Q型三种氮元素的相对含量分别约为56.4%、10.8%和32.7%. 这一结果也比之前一些工作27,31报道的要高, 这表明我们提供了一种制备高氮含量, 尤其是高N-6型氮含量纳米碳纤维的简便方法.
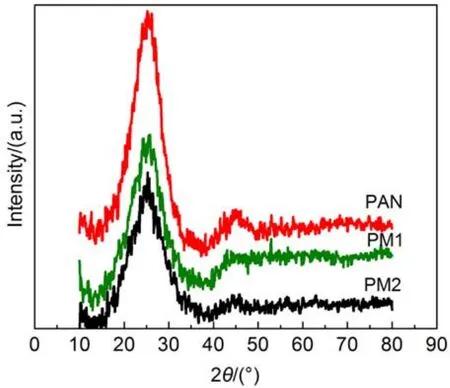
图6 PAN、PM1和PM2纳米碳纤维的XRD谱图Fig.6 XRD patterns of PAN, PM1, PM2 carbon nanofibers
3.4 纳米碳纤维的XRD及拉曼分析
图6是不同含量三聚氰胺改性的纳米碳纤维的XRD图谱. 从图中可以看出, 三种纳米碳纤维在2θ为25.5°、43.5°和53°左右都有衍射峰, 它们分别是石墨结构的 (002)衍射峰、(100)衍射峰和(004)衍射峰.32(002)衍射峰由石墨层堆叠产生, 相比之下三聚氰胺改性的两种纤维的(002)衍射峰均比较宽, 而且呈现不对称的峰形, 说明两种碳纤维中的石墨层沿c轴呈现乱层堆叠, 存在严重的晶格畸变. 三种碳纤维的(100)和(004)衍射峰均较低, 并且存在严重的宽化和交叠, 说明其类石墨的层状结构呈乱层堆叠状. 同时我们还看到, PAN纳米碳纤维的衍射峰较尖锐, 衍射峰强度高, 而三聚氰胺的加入导致衍射峰较低和衍射峰强度较弱, 说明三聚氰胺的加入使碳纤维类石墨层状结构的有序度降低, 类石墨结构的尺寸减小.
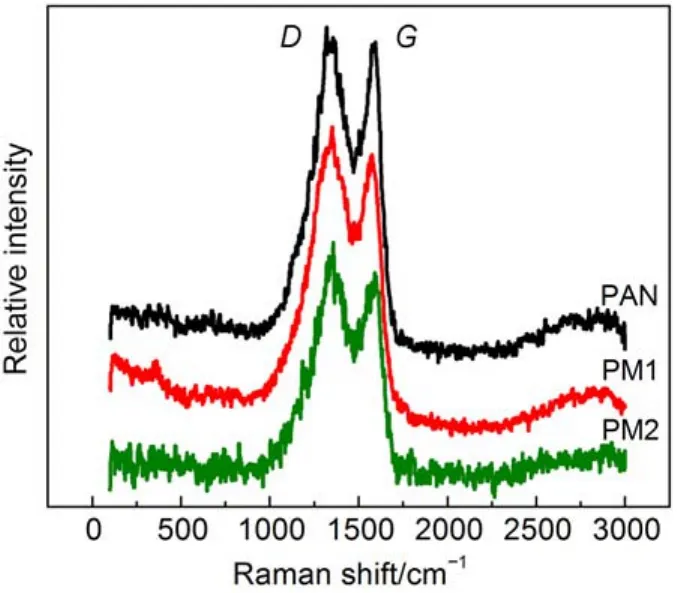
图7 PAN、PM1和PM2纳米碳纤维的拉曼图谱Fig.7 Raman spectra of PAN, PM1, and PM2 carbon nanofibers

表1 基于图7的D峰和G峰的拉曼位移Table1 Raman shift of D-peak and G-peak based on Fig.7
图7是三种纳米碳纤维的拉曼谱图. 从谱图中我们可以清楚看出, 在一级拉曼序区内, 碳纤维膜的拉曼光谱有两个明显的谱线: 1350 cm–1附近处D谱线和1580 cm–1附近处G谱线.33随着三聚氰胺含量的增加, 表征纳米碳纤维石墨结构中sp2杂化键结构完整程度的G线的强度逐渐减弱, 而表征石墨结构缺陷的sp3杂化键的D谱线则逐渐增强. 用ID和IG两者的相对强度比值ID/IG(R)来判断石墨化程度和石墨结构完整度, 由表1可知, 三种纳米碳纤维的R值分别为1.05、1.11和1.18, 这说明三聚氰胺的加入使得纳米碳纤维的石墨化程度降低, 结晶完整性下降,这与XRD测试结果相一致.
3.5 纳米碳纤维的比表面积和孔径分布
图8(A)为PAN、PM1和PM2纳米碳纤维样品的氮气吸附-脱附等温线. 可见, 该类曲线均为Langmuir第I类等温线, 说明PAN、PM1和PM2三种纳米碳纤维上主要分布的是微孔, 结合纳米纤维之间交联形成的各种孔径不一的介孔和大孔, 我们得到了具有多级孔道结构的三聚氰胺改性的纳米碳纤维电极材料. 相比PM1和PM2样品的吸附曲线, PAN纳米碳纤维的吸附量要小得多, 由多点BET法计算得到PAN、PM1和PM2纳米碳纤维样品的比表面积分别为212、461和428 m2g–1, 这一结果与我们的吸附量相互对应.

图8 PAN、PM1和PM2纳米碳纤维的吸脱附曲线图(A)和孔径分布图(B)Fig.8 Nitrogen adsorption-desorption isotherms (A) and pore size distributions (B) of PAN, PM1, and PM2 carbon nanofibers
图8B为PAN、PM1和PM2纳米碳纤维样品的孔径分布图, 可以看出, 孔径主要分布在5 nm以下,且绝大多数的孔为分布在2 nm以下的微孔, 同时PM1和PM2纳米碳纤维样品还具有一定含量的孔径在2–5 nm之间的介孔. 这些孔的形成可能一方面是由于预氧化过程中三聚氰胺熔融导致纳米有机纤维软化, 而与此同时三聚氰胺热分解产生气体, 以至这些气体在软化的有机纤维内部形成微孔并最终保持到纳米碳纤维内所致; 另一方面是由于在预氧化过程中PAN结构会产生变化而趋于稳定,34但是三聚氰胺的加入在一定程度上阻碍了这种结构的变化, 因此纳米碳纤维结构缺陷较多, 使纳米碳纤维具有更加丰富的微孔结构. 对比而言PAN纳米碳纤维上同样含有微孔但含量要少得多且没有介孔的存在, 这些孔径主要是由PAN纤维自身分解释放出气体产生的气孔所致. PM1样品的比表面积比PM2样品的略大, 是由于PM2样品中三聚氰胺的含量较高, 导致纤维之间交联的程度增大, 纤维交接点增多, 损失了部分面积. 由于三聚氰胺改性的纳米碳纤维具有合理的多级孔道结构, 加上较大的比表面积, 从而能够提高电极材料的比电容.
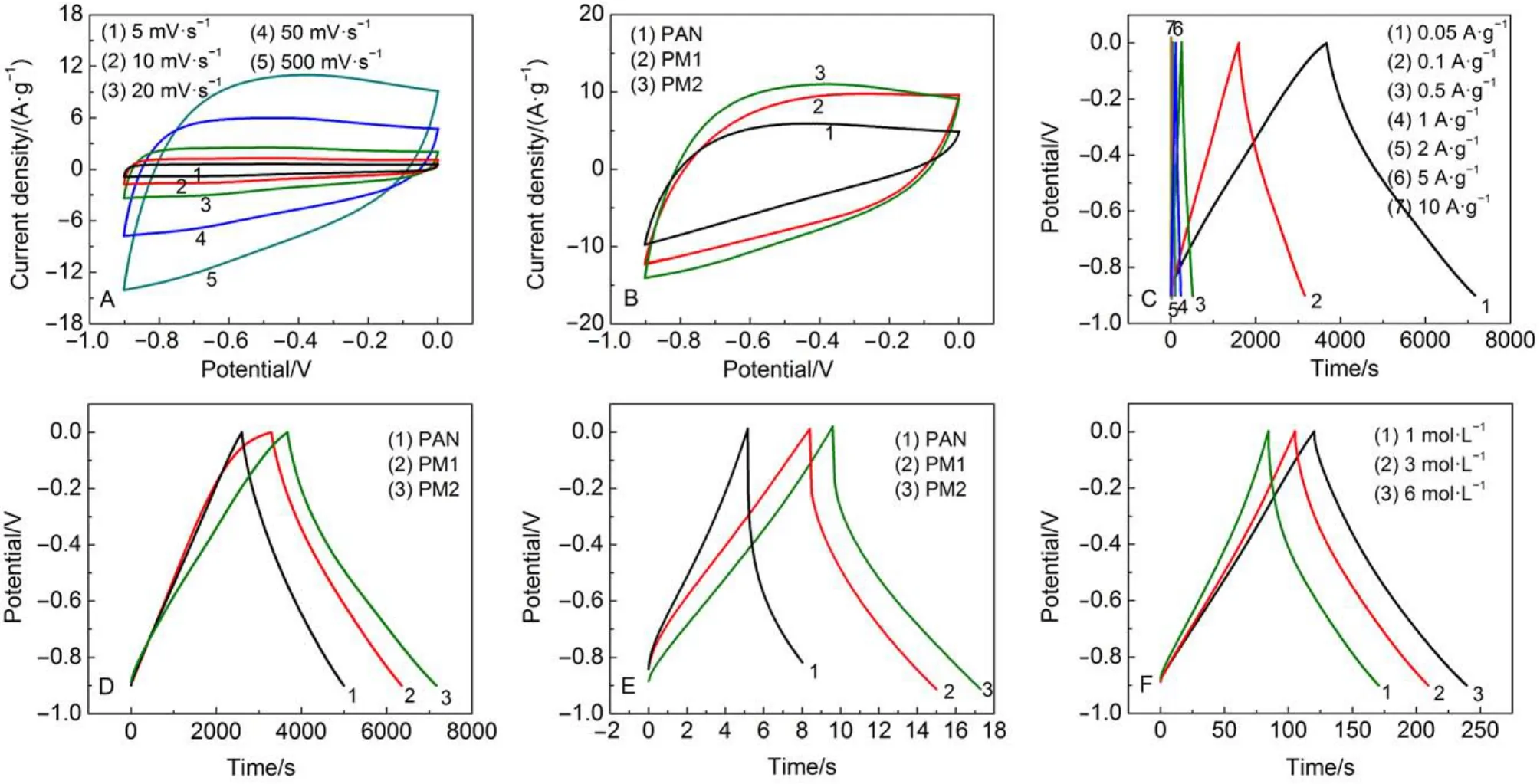
图9 (A)PM2纳米碳纤维电极在不同扫描速率下的CV曲线; (B) PAN、PM1和PM2纳米碳纤维电极在100 mVs–1扫描速率下的CV曲线; (C) PM2纳米碳纤维电极在不同电流密度下的恒流充放电曲线图; (D)和(E)分别为PAN、PM1和PM2纳米碳纤维电极在0.05和10 Ag–1电流密度下的恒流充放电曲线; (F) PM2纳米纤维电极在不同KOH浓度下的恒流充放电曲线Fig.9 (A) Cyclic voltammetry curves of PM2 carbon nanofiber electrode at different scan rates; (B) cyclic voltammetry curves of PAN, PM1, and PM2 carbon nanofiber electrodes at 100 mVs–1scan rate; (C) galvanostatic charge-discharge curves of PM2 carbon nanofiber electrode at different current densities; (D), (E) galvanostatic charge-discharge curves of PAN, PM1, and PM2 carbon nanofibers electrodes at different current densities ((D) 0.05 Ag–1, (E)10 Ag–1); (F) galvanostatic charge-discharge curves of PM2 carbon nanofiber electrode at different KOH concentrations
3.6 纳米碳纤维的电化学性能
从图9可见, 所有CV曲线均呈现双电层电容特性的类矩形特征, 但在小扫描速率下出现了部分的氧化还原峰, 这是由于聚丙烯腈本身含有的氰基以及三聚氰胺的加入使得纤维在碳化后仍保留部分的氮元素和含氮基团, 这些含氮基团不仅能够减小电解液与电极的接触电阻,35而且还能在电极测试时发生氧化还原反应而产生赝电容.36–39图9A为PM2纳米碳纤维电极在不同扫描速率下的CV曲线.从图中可以发现, PM2纳米碳纤维电极在较大扫描速率下仍能维持类矩形特征. 图9B为三种不同纳米纤维电极在100 mVs–1扫描速率下的CV曲线. 从图中可以发现, PAN纳米碳纤维电极变形严重, 而三聚氰胺改性的纳米碳纤维电极仍能维持类矩形的特征, 说明三聚氰胺改性的三维网络纳米碳纤维电极在大扫描速率下仍能维持较高的比电容和良好的速率特性.
图9C为PM2纳米碳纤维电极在不同电流密度下的恒流放电曲线. 恒流充放电实验在–0.9―0 V之间进行测试, 略带弧形的平滑曲线主要是由于法拉第赝电容反应引起的,27表明PM2纳米碳纤维表现出法拉第赝电容. 图9D、9E为三组电极在0. 05和10 Ag–1电流密度下的放电曲线对比图, PM1和PM2曲线有一定程度的弯曲, 表现出部分的赝电容, 但曲线仍主要为对称的三角形双电层特性. 从图中可以看出, PM2电极的放电时间最长, 说明PM2电极具有最大的比电容值, 其值高达194 Fg–1. 在10 Ag–1电流密度下, 相比于PM2电极, PAN电极内阻较大, 在放电开始时电压降比较大, 比电容值衰减严重. PAN电极与PM2电极相比具有较大的内阻, 这与SEM照片的形貌特性相印证. 图9F为PM2电极在1、3和6 molL–1的KOH电解质溶液中, 1 Ag–1电流密度下的恒流充放电曲线. 随着电解质溶液浓度的增大, 相应的比电容依次增大, 电压降则相应地减小, 这主要是因为电解液浓度增大, 电导率增加, 减小了电容器的内阻, 有利于大电流下的电容器比电容的提高.40
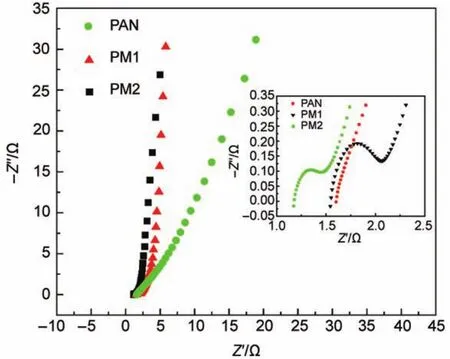
图10 PAN、PM1和PM2纳米碳纤维电极的电化学阻抗谱(EIS)曲线Fig.10 Electrochemical impedance spectroscopy (EIS) curves of PAN, PM1, and PM2 carbon nanofiber electrodes
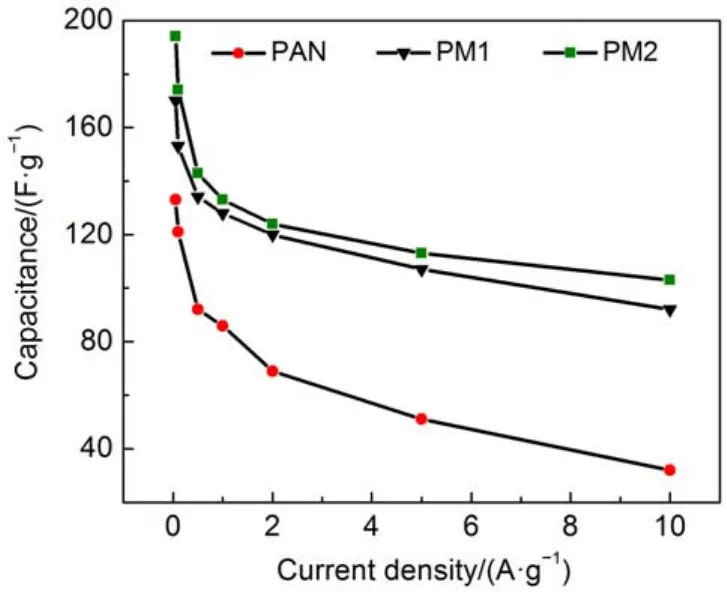
图11 三种纳米碳纤维电极在不同电流密度下的比电容值Fig.11 Specific capacitance of three carbon nanofiber electrodes at various current densities
图10是PAN、PM1和PM2三种纳米碳纤维电极样品分别在三电极体系下的EIS谱. 与PAN电极相比, 三聚氰胺改性的电极曲线在低频的斜率更大,且随着电极中三聚氰胺比例的增大, 低频区的斜率逐渐增大, 表明电极与电解液之间的传质速率随着三聚氰胺含量的增多而不断上升. 这主要是因为三聚氰胺的加入增加纳米碳纤维三维网络的搭接程度提高, 并且含氮基团数目增多, 改善了电极的亲水性, 使得电极与电解液之间的电阻减小, 有利于电子的快速扩散. 图11是三组电极在不同电流密度下的比电容值. 从图中可以发现, PM2在0.05 Ag–1时的比电容值最大, 最大值为194 Fg–1; 在10 Ag–1时比电容值为104 Fg–1, 保持率为53.6%. 而PAN纳米碳纤维在0.05 Ag–1时的比电容值为133 Fg–1; 在10 Ag–1时比电容值仅32 Fg–1, 保持率仅24.1%. 这表明, 三聚氰胺改性纳米碳纤维电极较PAN纳米碳纤维具有良好的倍率特性.

图12 PAN、PM1和PM2纳米碳纤维电极在2 Ag–1电流密度下的循环稳定性Fig.12 Cycling stability of PAN, PM1, and PM2 carbon nanofiber electrodes at a current density of 2 Ag–1
图12是PAN、PM1和PM2三种纳米碳纤维电极在电流密度为2 Ag–1时循环充放电1000次的比电容值变化曲线. 经计算, PM2电极的首次充放电比电容值为124 Fg–1, 第1000次充放电时其比电容值为123 Fg–1, 保持率为99.2%, 这说明三聚氰胺改性纳米碳纤维优异的电化学稳定性.
4 结 论
采用聚丙烯腈和三聚氰胺为原料, 通过静电纺丝法我们成功制备了含有三聚氰胺的有机纳米纤维, 经过预氧化和碳化工艺制备得到了多孔纳米碳纤维并将其用作超级电容器电极材料. 三聚氰胺改性的纳米碳纤维具有较高的电导率, 较细的直径和较大的比表面积, 在未经活化时其比电容值可以达到194 Fg–1, 在2 Ag–1电流密度下循环充放电1000次后比电容保持率高达99.2%, 体现了较好的电容稳定特性. 通过三聚氰胺改性制备的有机纳米碳纤维和碳化后的交联网络结构的纳米碳纤维不仅在电容器电极材料, 而且可望在其他领域具有良好的潜在应用前景.
(1)Li, Z. H.; Li, S. J.; Zhou, J.; Zhu, T. T.; Shen, H. L.; Zhuo, S. P. Acta Phys. -Chim. Sin. 2015, 31 (4), 676. [李朝辉, 李仕蛟,周 晋, 朱婷婷, 沈红龙, 禚淑萍. 物理化学学报, 2015, 31(4), 676.] doi: 10.3866/PKU.WHXB201501281
(2)Li, L.; He, Y. Q.; Chu, X. F.; Li, Y. M.; Sun, F. F.; Huang, H. Z. Acta Phys. -Chim. Sin. 2013, 29 (8), 1681. [李 乐, 贺蕴秋, 储晓菲, 李一鸣, 孙芳芳, 黄河周. 物理化学学报, 2013, 29 (8), 1681.] doi: 10.3866/PKU.WHXB201305223
(3)Xu, G. Y.; Ding, B.; Nie, P.; Luo, H. J.; Zhang, X. G. Acta Phys. -Chim. Sin. 2013, 29 (3), 546. [徐桂银, 丁 兵, 聂 平,骆宏钧, 张校刚. 物理化学学报, 2013, 29 (3), 546.] doi: 10.3866/PKU. WHXB201301081
(4)Chen, C. J.; Hu, Z. A.; Hu, Y. Y.; Li, L.; Yang, Y. Y.; An, N.; Li, Z. M.; Wu, H. Y. Acta Phys. -Chim. Sin. 2014, 30 (12), 2256. [陈婵娟, 胡中爱, 胡英瑛, 李 丽, 杨玉英, 安 宁, 李志敏, 吴红英. 物理化学学报, 2014, 30 (12), 2256.] doi: 10.3866/PKU. WHXB201409302
(5)Guo, P. Z.; Ji, Q. Q.; Zhang, L. L.; Zhao, S. Y.; Zhao, X. S. Acta Phys. -Chim. Sin. 2011, 27 (12), 2836. [郭培志, 季倩倩, 张丽莉,赵善玉, 赵修松. 物理化学学报, 2011, 27 (12), 2836.] doi: 10.3866/PKU.WHXB20112836
(6)Ma, G. F.; Mu, J. J.; Zhang, Z. G.; Sun, K. J.; Peng, H.; Lei, Z. Q. Acta Phys. -Chim. Sin. 2013, 29 (11), 2385. [马国富, 牟晶晶,张稚国, 孙看军, 彭 辉, 雷自强. 物理化学学报, 2013, 29 (11), 2385.] doi: 10.3866/PKU.WHXB201309051
(7)Wang, J. D.; Peng, T. J.; Sun, H. J.; Hou, Y. D. Acta Phys. -Chim. Sin. 2014, 30 (11), 2077. [汪建德, 彭同江, 孙红娟, 侯云丹. 物理化学学报, 2014, 30 (11), 2077.] doi: 10.3866/PKU.WHXB 201409152
(8)Peng, X.; Li, D. Q.; Peng, J.; Peng, L. L.; Wu, C. Z.; Xie, Y. Bull Sci. Technol. 2013, 58 (28–29), 2886. [彭 旭, 李典奇, 彭 晶,彭乐乐, 吴长征, 谢 毅. 科学通报, 2013, 58 (28–29), 2886.]
(9)Lai, C.; Zhou, Z.; Zhang, L.; Wang, X.; Zhou, Q.; Zhao, Y.; Wang, Y.; Wu, X. F.; Zhu, Z.; Fong, H. J. Power Sources 2014, 247, 134. doi: 10.1016/j.jpowsour.2013.08.082
(10)Yang, S.; Xu, G. Y.; Han, J. P.; Bing, H.; Dou, H.; Zhang, X. G. Acta Phys. -Chim. Sin. 2015, 31 (4), 685. [杨 硕, 徐桂银, 韩金鹏, 邴 欢, 窦 辉, 张校刚. 物理化学学报, 2015, 31 (4), 685.] doi: 10.3866/PKU.WHXB201502022
(11)Zhao, L.; Wu, Q.; Han, R.; Wu, J.; Yao, W. Materials Review 2013, 27 (11), 54
(12)Su, D. S.; Chen, X.; Weinberg, G.; Klein-Hofmann, A.; Timpe, O.; Hamid, S. B. A.; Schlögl, R. Angew. Chem. Int. Edit. 2005, 44 (34), 5488.
(13)Sharma, S.; Pollet, B. G. J. Power Sources 2012, 208, 96. doi: 10.1016/j.jpowsour.2012.02.011
(14)Wu, C.; Yao, X.; Zhang, H. Int. J. Hydrog. Energy 2010, 35 (1), 247. doi: 10.1016/j.ijhydene.2009.10.079
(15)Chinthaginjala, J.; Seshan, K.; Lefferts, L. Ind. Eng. Chem. Res. 2007, 46 (12), 3968. doi: 10.1021/ie061394r
(16)Huang, Z. M.; Zhang, Y. Z.; Kotaki, M.; Ramakrishna, S. Compos. Sci. Technol. 2003, 63 (15), 2223. doi: 10.1016/S0266-3538(03)00178-7
(17)Jung, M. J.; Jeong, E.; Kim, Y.; Lee, Y. S. J. Ind. Eng. Chem. 2013, 19 (4), 1315 doi: 10.1016/j.jiec.2012.12.034
(18)Kim, B. H.; Yang, K. S. J. Electroanal. Chem. 2014, 714, 92.
(19)Park, G. S.; Lee, J. S.; Kim, S. T.; Park, S.; Cho, J. J. Power Sources 2013, 243, 267. doi: 10.1016/j.jpowsour.2013.06.025
(20)Hang, Z. S.; Tan, L. H.; Cao, X. M.; Ju, F. Y.; Ying, S. J.; Xu, F. M. Mater. Lett. 2011, 65 (7), 1079. doi: 10.1016/j.matlet. 2011.01.010
(21)Kim, B. H.; Yang, K. S. J. Ind. Eng. Chem. 2014, 20 (5), 3474. doi: 10.1016/j.jiec.2013.12.037
(22)Hang, Z. S.; Tan, L. H.; Ju, F. Y.; Zhou, B.; Ying, S. J. J. Anal. Sci. 2011, 27 (3), 279. [杭祖圣, 谈玲华, 居法银, 周 斌, 应三九. 分析科学学报, 2011, 27 (3), 279.]
(23)Yuan, B.; Zheng, X. Y.; Zhang, C.; Lu, W.; Li, B. H.; Yang, Q. H. New Carbon Materials 2014, 29 (6), 426. doi: 10.1016/S1872-5805(14)60147-5
(24)Wu, H.; Hu, L.; Rowell, M. W.; Kong, D.; Cha, J. J.; McDonough, J. R.; Zhu, J.; Yang, Y.; McGehee, M. D.; Cui, Y. Nano Lett. 2010, 10 (10), 4242. doi: 10.1021/nl102725k
(25)Qiu, Y. J.; Yu, J.; Shi, T. G.; Zhou, X. S.; Bai, X. D.; Huang, J. Y. J. Power Sources 2011, 196, 9862. doi: 10.1016/j.jpowsour.2011.08.013
(26)Sun, L.; Tian, C. G.; Fu, Y.; Yang, Y.; Yin, J.; Wang, L.; Fu, H. G. Chem. Eur. J. 2014, 20, 564. doi: 10.1002/chem.v20.2
(27)Nan, D.; Huang, Z. H.; Lv, R. T.; Yang, L.; Wang, J. G.; Shen, W. C.; Lin, Y. X.; Yu, X. L.; Ye, L.; Sun, H. Y.; Kang, F. Y. J. Mater. Chem. A 2014, 2, 19678. doi: 10.1039/C4TA03868A
(28)Li, M.; Xue, J. J. Phys. Chem. C 2014, 118 (5), 2507. doi: 10.1021/jp410198r
(29)Muhammad, T.; Chuan, B. C.; Nasir, M.; Faheem, K. B.; Asif, M.; Faryal, I.; Sajad, H.; M, T.; Zulfiqar, A.; Imran, A. ACS Appl. Mater. Interfaces 2014, 6, 1258.
(30)Ya, M.; Hui, D.; Bin, X.; Lin, Z.; Yong, S. H.; Chang, C. X. Energy Environ. Sci. 2012, 5 (7), 7950.
(31)Chuan, G. H.; Qing, H.; Fei, Z.; Zi, Y. Y.; Nan, C.; Liang, T. Q. Nanoscale 2013, 5, 2726. doi: 10.1039/c3nr34002c
(32)An, G. H.; Ahn, H. J. Carbon 2013, 65, 87. doi: 10.1016/j.carbon.2013.08.002
(33)Wang, Y.; Serrano, S.; Santiago-Aviles, J. J. Synthetic Metals 2003, 138 (3), 423. doi: 10.1016/S0379-6779(02)00472-1
(34)Lu, J. W.; Ren, X. Z.; Chen, Y. Z.; Dong, M.; Zhang, Z. P.; Yu,J.; Guo, C. Y. Chem. J. Chin. Univ. 2008, 29 (9), 1870. [陆建巍,任祥忠, 陈艺章, 董 穆, 张展鹏, 于 建, 郭朝霞. 高等学校化学学报, 2008, 29 (9), 1870.]
(35)Yang, X.; Wu, D.; Chen, X.; Fu, R. J. Phys. Chem. C 2010, 114 (18), 8581. doi: 10.1021/jp101255d
(36)Su, F.; Poh, C. K.; Chen, J. S.; Xu, G.; Wang, D.; Li, Q.; Lin, J.; Lou, X. W. Energy Environ. Sci. 2011, 4 (3), 717. doi: 10.1039/C0EE00277A
(37)Wen, Z.; Wang, X.; Mao, S.; Bo, Z.; Kim, H.; Cui, S.; Lu, G.; Feng, X.; Chen, J. Adv. Mater. 2012, 24 (41), 5610. doi: 10.1002/adma.201201920
(38)Xiao, N.; Lau, D.; Shi, W.; Zhu, J.; Dong, X.; Hng, H. H.; Yan, Q. Carbon 2013, 57, 184. doi: 10.1016/j.carbon.2013.01.062
(39)Hassan, F. M.; Chabot, V.; Li, J.; Kim, B. K.; Ricardez-Sandoval, L.; Yu, A. J. Mater. Chem. A 2013, 1 (8), 2904. doi: 10.1039/c2ta01064j
(40)Li, Z. P.; Zhao, J. G.; Wen, Y. Q.; Li, J.; Xing, B. Y.; Guo, Y. Chem. Ind. Eng. Prog. 2012, 31 (8), 1631. [李作鹏, 赵建国, 温雅琼, 李 江, 邢宝岩, 郭 永. 化工进展, 2012, 31 (8), 1631.]
欢迎订阅《大学化学》
经北京市新闻出版广电局批复(京新广函[2015]200号),自2016年1月起:《大学化学》(CN11-1815/O6)由双月刊变更为月刊。
《大学化学》是由教育部主管、北京大学和中国化学会共同主办的化学教育类刊物。主要介绍化学科学的新进展,开展与教学有关的重大课题的研讨,交流教学改革经验,报道化学及其相关学科的新知识、新动向,促进教师知识更新,扩大学生知识面,为提高教学水平服务。主要栏目有:今日化学、教学研究与改革、知识介绍、化学实验、师生笔谈、自学之友、大学化学先修课程、竞赛园地、国外化学教育、化学史、书评以及专题讨论等。
2016年每本定价12.00元,全年出版12期,共144.00元。
全国各地邮局均可订阅,邮发代号:82-314。为方便读者订阅,本刊编辑部全年办理邮购业务。
邮购地址:北京大学化学学院 《大学化学》编辑部
邮编:100871
电话:010-62751721
Email:dxhx@pku.edu.cn
网址:http://www.dxhx.pku.edu.cn
《大学化学》编辑部
Preparation of Cross-Linked Porous Carbon Nanofiber Networks by Electrospinning Method and Their Electrochemical Capacitive Behaviors
LU Jian-Jian YING Zong-Rong*LIU Xin-Dong ZHAO Shuang-Sheng
(School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, P. R. China)
Cross-linked porous carbon nanofiber networks were successfully prepared by electrospinning followed by preoxidation and carbonization using low-cost melamine and polyacrylonitrile (PAN) as precursors. The structures and morphologies of the nanofiber networks were investigated using Fourier-transform infrared (FTIR) spectroscopy, thermogravimetric analysis (TGA), scanning electron microscopy (SEM), X-ray diffraction (XRD), Raman spectroscopy, and N2adsorption/desorption. The carbon fibers had an interconnected nanofibrous morphology with a well-developed porous structure including micropores, mesopores and macropores, high-level nitrogen doping (up to 14.3%), and a small average diameter (about 89 nm). Without activation, the carbon nanofibers had a high specific capacitance of 194 Fg–1at a current density of 0.05 Ag–1. Cycling experiments showed that the specific capacitance retained approximately 99.2% of the initial capacitance after 1000 cycles at a current density of 2 Ag–1, indicating an excellent electrochemical performance.
Melamine; Electrospinning; Carbon nanofiber; Cross-linked network; Supercapacitor
©Editorial office of Acta Physico-Chimica Sinica
O646
10.3866/PKU.WHXB201510081
Received: May 19, 2015; Revised: October 8, 2015; Published on Web: October 8, 2015.*
