尖晶石锰酸锂电池容量衰减的机理探析
2015-12-05陈海辉邱新平
陈海辉,邱新平
尖晶石锰酸锂电池容量衰减的机理探析
*陈海辉1,2,邱新平2
(1.井冈山大学化学化工学院,江西,吉安 343009;2.清华大学化学系,北京100084)
尖晶石型锰酸锂的容量衰减是限制其大规模应用的瓶颈问题。现有观点认为二价锰离子在阳极上还原成金属锰,催化电解质分解、毒化固体电解质界面(SEI)膜,造成了锰酸锂体系锂离子电池的容量衰减。但最新研究确定锰元素是以二价的锰沉积在阳极上,并没有被还原成金属锰。本研究介绍国内外锰系锂离子电池容量衰减的研究进展,并在“沉积阳极上的锰氧化态是+2价”这个新发现的基础上,结合分析其它文献的实验数据,归纳总结出 “二价锰离子与阳极SEI膜中的活性锂离子进行离子交换而沉积在阳极上,阻碍、堵塞活性锂离子的自由进出,从而引起锂离子电池容量衰退”新观点。
容量衰减;锰沉积;锰酸锂;锂离子电池
锂离子电池作为绿色能源,越来越受到各国政府的重视,人们期望锂离子电池有更长的寿命、更大的容量和更高的安全性[1],延长锂离子电池的循环寿命是现阶段的研究重点之一[2]。
尖晶石型锰酸锂LiMn2O4具有资源丰富、能量密度高、成本低、无污染、安全性好等优点,被人们认为是锂离子动力电池理想的正极材料[3-5]。但是LiMn2O4系锂离子电池的容量衰减较快,尤其在高温条件下,一直是限制其大规模应用的瓶颈[6-7]。
通过掺杂金属离子[1-4,8]、材料纳米化[9-10]稳定LiMn2O4的尖晶石结构,或者通过表面包覆减少锰的溶解[5-6,11],或在电解质中加添加剂[12]抑制锰在阳极上的沉积,尖晶石锰酸锂系锂离子电池的循环性能得到了一些改善,但容量损失依然严重[13]。
对于锰酸锂来说,锰溶解一直被认为是引起电池容量衰减最主要的原因之一[13-15]。现有理论认为尖晶石锰酸锂发生歧化反应[16-20],可溶解的Mn2+从阴极迁移到阳极上,Mn2+在阳极表面上还原成金属锰,阳极SEI膜被金属锰毒化[21],金属锰催化电解质分解[22-23],从而造成了锰酸锂体系的电池容量衰减。
一般认为Mn(II)在阳极的低电位下被电化学还原成金属锰[23-24]、或者被锂化石墨化学还原成金属锰[22,25]。2013年10月,清华大学邱新平研究团队采用先进的表征技术(XAENS、XPS)确凿地证实了沉积在阳极上的锰是以“+2价氧化态”存在[26],与传统文献推测(金属态)显然不同。该重要研究成果发表在国际著名学术期刊Nature Comm.上。本研究在此基础上,展现国内外锰系锂离子电池容量衰减的研究进展,分析已有文献的实验数据,提出新机理。
1 国内外研究进展
锂离子电池的充放电过程是一个复杂的电化学过程,导致电池容量衰减的因素也不是单一的。目前人们正在从正极材料结构变化、正极材料溶解、锂离子消耗、内阻增加等方面来阐述锰酸锂电池的容量衰减原因:
1.1 正极材料结构变化
正极材料由于相转移和体相结构的变化引起的不可逆变化对电池寿命都有很大的影响[27-28]。LiMn2O4在放电末期发生Jahn-Teller效应,破坏材料结构引起容量衰减[29]。
1.2 正极材料溶解
正极材料溶解是导致电池容量衰减的原因之一[30-31]。在尖晶石型 LiMn2O4体系电池中,锰溶解是造成电池容量衰减的一个重要原因[32]。Mn2+和电解液的分解产物形成氟化物或者氧化物沉积到电极表面[31],导致电极的阻抗增加,堵塞锂离子扩散通道,引起容量的衰减。
1.3 锂离子消耗
储存在高温下电池的容量衰减主要原因是石墨阳极与电解液发生界面反应,导致可循环锂离子减少造成的[33]。锰酸锂体系溶解的 Mn2+迁移到阳极并沉积在其表面,破坏了 SEI 膜或者阳极表面形貌,从而影响阳极性能[33]。
1.4 内阻增加
引起内阻增加主要来自两个方面:阳极 SEI 膜不稳定,不断在表面形成新的表面膜使得极化增加,电池内阻增加[34-36]。在碳阳极表面形成 SEI 膜不仅消耗了锂离子,还增加了阳极的阻抗所以导致了电池倍率性能变差。
这些研究可以总结为:尖晶石锰酸锂阴极在电化学过程中溶解加剧溶解,不仅减少活性正极材料的量,还会引起正极结构的变化,而更严重的是溶解金属离子迁移至负极表面、沉积在负极表面,影响了阳极SEI 膜的稳定性。破损的阳极SEI膜,造成消耗锂离子、分解电解液,还造成负极阻抗增大、电池内阻增加,从而严重影响电池性能。
通常认为由于HF酸的作用,锰从阴极溶解、迁移到阳极上沉积导致严重的电池容量衰减[12]。现在发现有些过渡金属,如LiNi1/3Co1/3Mn1/3O2(NCM)[14]、LiFePO4[37],也导致了锂离子电池容量衰减。研究过渡金属锰的沉积过程,及其导致锂离子电池容量衰减的机理已成为共性、关键问题,这是当今锂离子电池研究的发展趋势。
现有理论认为锰溶解、迁移、沉积过程的机制为:(1) 尖晶石锰酸锂LiMn2O4中的Mn(III)在阴极和电解液之间的界面处发生歧化反应[16-20],。Mn2+物种溶解在电解质中,Mn4+物种不溶解电解液而富集在阴极上;(2)可溶解的Mn2+经过扩散、电化学过程,从阴极迁移到阳极上; (3) Mn2+在阳极表面上还原成金属锰,。有人认为Mn(II)在阳极的低电位下被电化学还原成金属锰[21]、有人认为被锂化石墨化学还原成金属锰[22],但都推测出DMD的第三步:Mn2+在阳极表面上还原成金属锰[21-23,38-39]。虽然有2篇文献[40-41]经X射线光电子谱(XPS)测量,表明在石墨阳极上沉积了Mn2+粒子,但2篇文献的作者认为Mn2+首先还原成金属锰而沉积在阳极表面上,后来才氧化成Mn2+。这2篇文献[40-41]的看法与上描述的第三步类似。基于以上沉积反应机制,人们认为金属锰沉积使石墨阳极的SEI结构发生改变或分解[21-23]是导致电池容量衰减的直接原因。
清华大学邱新平教授团队在研究阳极上锰的存在价态有重大发现,2013年10月在Nature Comm.上发表文章:首次确定锰元素是以二价的锰(氧化物)沉积在阳极上,并没有被还原成金属锰[26]。样品的Mn-O键长都在(2.15 ± 0.03) Å[26],比MnO的键长2.222 Å稍短。在电池循环实验中,LMO经过修饰,电池体系AC阻抗的增加量,比未修饰的小多了[26]。
通过表征SEI的结构、组分来研究锂离子电池性能已经成为一种研究方法。用参比电极电池体系,测量锂离子电池阳极和阴极在循环过程中的AC阻抗,已成为一种便捷的原位测试技术[43]。与XANES技术有所不同,XPS对表面更敏感(测量深度1-3 nm),因此是用于分析阳极表面上SEI膜的理想工具[44]。原位FTIR直接确定发生在阳极上的反应,了解阳极SEI膜的组成成分、锰化合物[45]。原位TEM可以在线表征循环过程中SEI的结构变化[46]。采用先进的表征技术研究锂离子电池SEI的结构和化学组分已经成为一种趋势。
目前理论还难以回答我们发现的一个新现象:四种阳极(石墨、钛酸锂、磷酸铁锂、金属锂)与尖晶石锰酸锂组装成锂离子电池,即使在阳极上沉积了同样多量的Mn2+,但它们组成的锂离子电池的容量衰减情况却有显著差别。我们发现石墨或金属锂组成的锂离子电池的容量衰减量随沉积Mn2+量增加而增加,而钛酸锂、去锂磷酸铁锂组成的电池容量衰减在55 ℃下并没有随沉积Mn2+的量增加而增加[36],但有文献表明:钛酸锂/尖晶石锰酸锂电池体系,在80 ℃以后却表现出剧烈衰减现象[42]。
2 实验数据分析
2.1 不同的阳极容量衰减对比
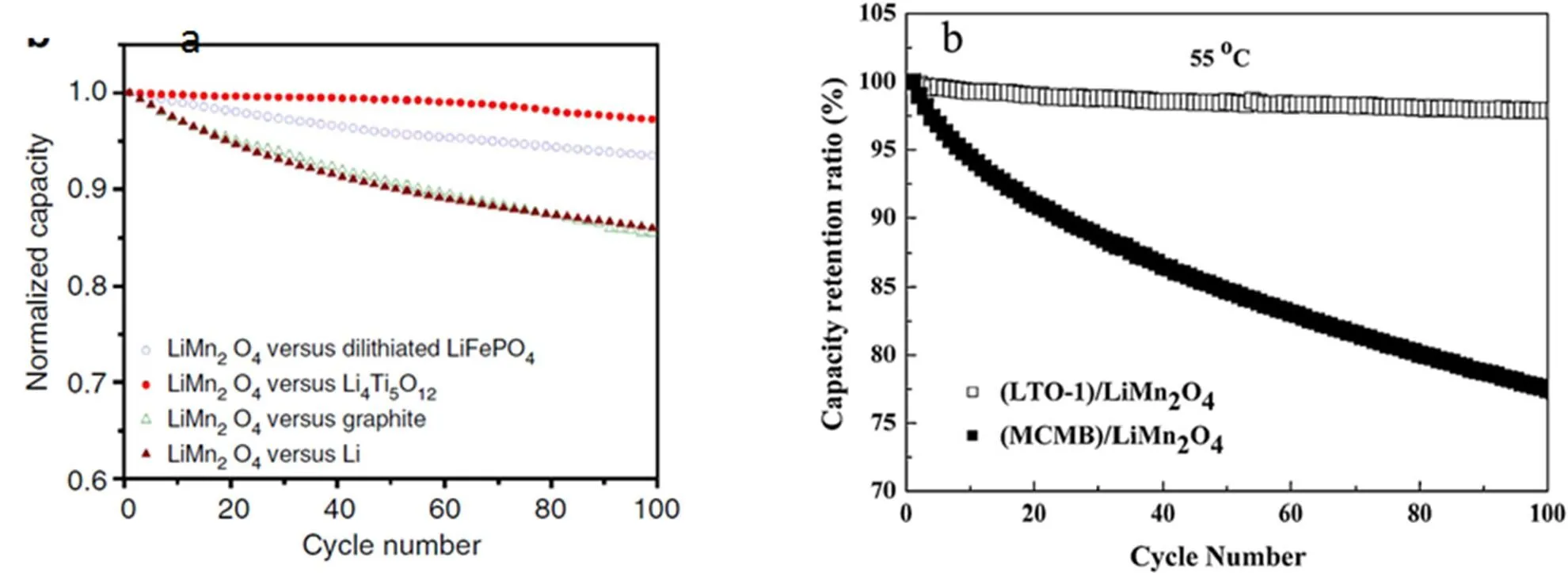
图1 对应不同阳极锰酸锂电池的容量衰减率
锰酸锂与不同阳极材料所组装的元电池在工作环境55℃容量衰减情况如图1。在1(a)中阳极材料分别为LiFePO4、Li4TI5O12、石墨、金属,经过100次循环之后,LiMn2O4/Li4Ti5O12(LTO) 锂离子电池几乎没有衰减,LiMn2O4/LiFePO4(D-LFP) 锂离子电池衰减5% LiMn2O4/MCMB容量衰减却高达15%,而经过计算由于阴极活性物质Mn3+的流失造成的容量衰减量< 1%[26]。在图1(b)中阳极材料为Li4TI5O12(LTO) 和微球石墨(MCMB),容量衰减情况与1(a)的相近[47]。从这些实验中,我们得出:与阳极相比,阴极导致的容量衰减几乎是可以忽略,锰系锂离子电池的容量衰减主要是由于阳极衰减导致。
2.2 锰沉积量的影响

图2 电极浸泡后性能

图3 循环性能及ICP-AAS测试
图2表明石墨电极在25℃含二价锰离子的电解液中浸泡2 d,与金属锂组装成原电池的电化学循环性能,结果表明随着锰离子沉积量越多,其容量衰减也就越快。锰沉积开始较快,然后变缓慢,容量衰减率也是先快后慢[48]。
图3是Li/LiMn2O4半电池的循环性能曲线(黑色曲线)和锰离子在Li阳极SEI层的锰沉积量(红色曲线)。在100次循环之后,锰离子浓度达400 p.p.m.。试验显示:100次循环,沉积在MCMB阳极上的锰量为320 p.p.m.,D-LFP阳极上260 p.p.m.,LTO阳极上300 p.p.m.。虽然在100次循环之后,LTO和D-LFP上即使锰沉积量一样多,但却容量衰减却很少[8]。这些表现可以帮我们更好地理解不同阳极的容量衰减表现差别,这也暗示了MCMB-LMO原电池的容量衰减并不是活性物质Mn3+的流失直接导致,可能是电解液中的Mn2+与石墨SEI层发生反应,改变了得SEI层的性质[26]。
温度和石墨锂化状态对容量衰减的影响如图[48]。如前所述,石墨在含有二价锰离子电解液中浸泡时间越长,工作环境温度高,在阳极上锰沉积量就越多,其容量衰减较快。但图4中有一个特别现象:石墨在冲锂状态比去锂状态下,其容量衰减更快。这个特别现象表明:二价锰更容易沉积在冲锂状态下石墨上,即石墨中锂离子越多,锰离子就越容易沉积。该现象暗示了锰离子与锂离子进行了交换吸附。
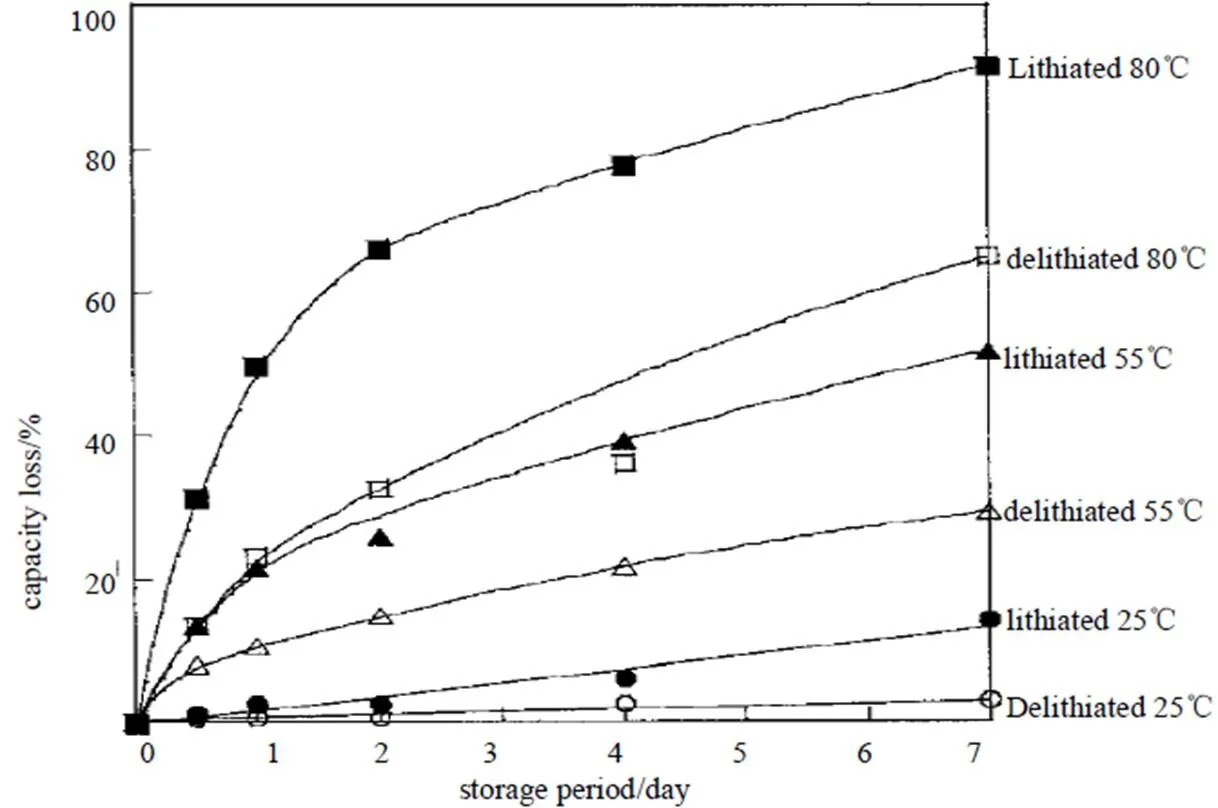
图4 温度和锂化状态对循环性能的影响
2. 3 SEI的形态
沉积锰能改变石墨SEI层的结构或组分,导致电池容量衰减。图5为石墨阳极在循环前后的微米级SEM照片对比,清晰地表明了在循环之后的石墨表面上出现了微小的颗粒[49]。

图5 石墨阳极的表面形态
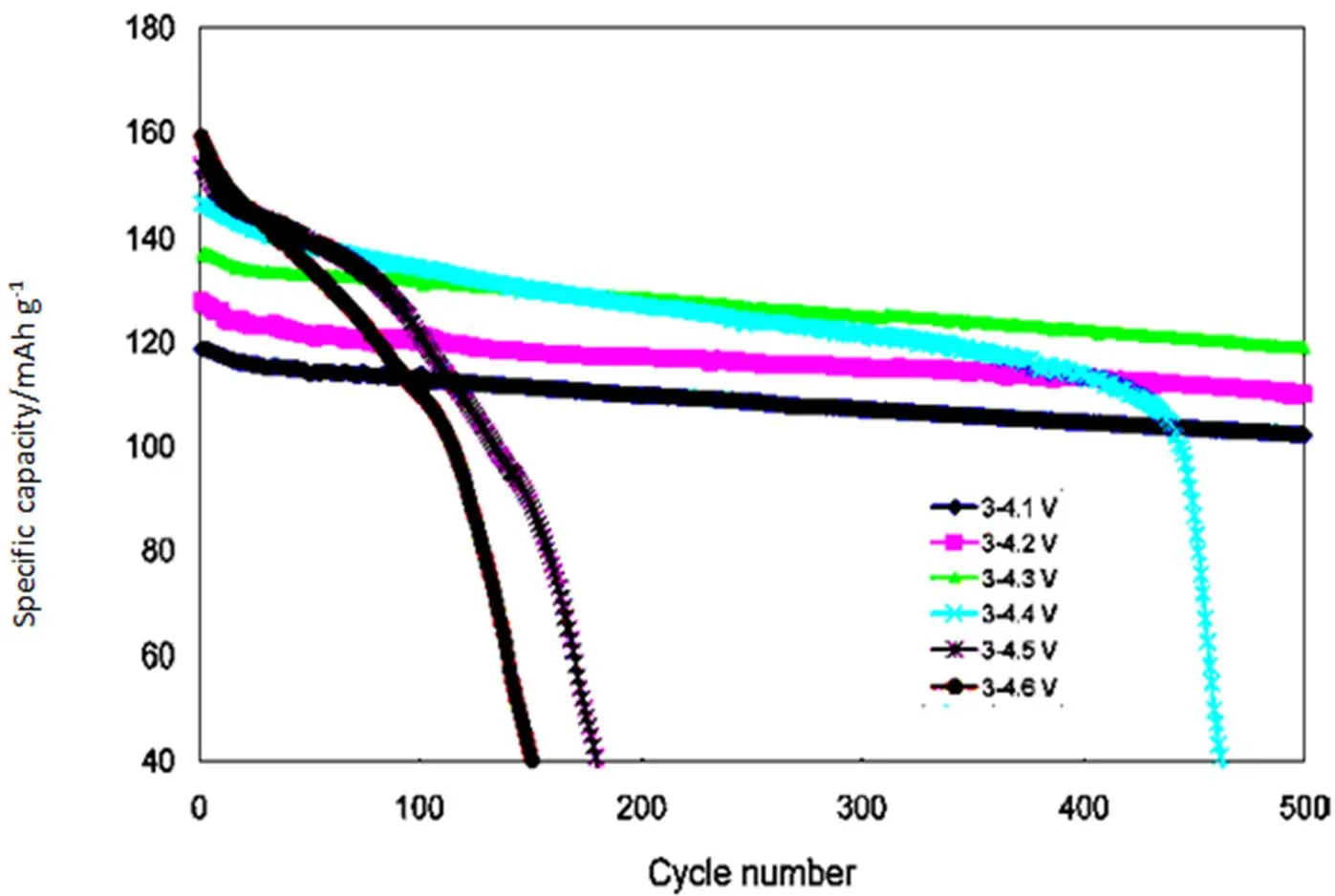
图6 过冲循环性能
NCM /MCMB电池体系循环性能如图6,过冲电压影响非常大,4.5 V电压下充电,容量急剧衰减[15]。对应的石墨阳极表面形态如图7,在4.3V电压下充电下的循环之后,与循环之前相比,MCMB颗粒外观变大表表明在表面覆盖了一层较厚实的物质即形成SEI膜层[15];在4.5 V电压下充电下的循环之后,与循环之前相比,MCMB表面除了形成SEI膜层,还出现了许多更小颗粒[15]。
这些小颗粒的EDX如图8,主要元素是C,O,F,P. O,还有Mn, Ni, Co(来源于溶解电极), Al(来源于集流片)元素。这个SEM图清晰表面在电解液中的Mn2+与石墨的SEI发生反应,改变了SEI性能,导致了容量衰减[15]。

图8 循环后MCMB阳极的EDX结果
2. 4 充分电倍率影响
NCM阴极与微球石墨(MCMB)阳极循环前后在不同倍率下的容量变化情况如图9:在1 C倍率冲放电下,循环后微球石墨(MCMB)阳极组装的电池其容量损失达80%。全电池失效是由于阳极的失效[15]。结果显示石墨的电化学性能主要是受溶解在电解液中的过渡金属离子影响。

图9 MCMB循环之后的倍率表现

图10 1C 和20C倍率的循环表现
LiMn2O4/ Li4Ti5O12全电池在1C和20C倍率冲放电下,电化学循环性能如图10:1C倍率下,经过500次循环,容量依然保持在97%;在20C倍率下,经过2000次循环,容量依然保持在80%[47]。
图9-图10暗示了在SEI中锂离子进出通道越多,倍率就可以越高,容量维持也越高。相反,与新制作的没有经过循环充分电的相比,循环后的微球石墨(MCMB)阳极的SEI中锂离子进出通道就比较少了,所以容量衰减也就较多,即经过电化学循环之后,SEI中锂离子进出通道被堵塞了一些。
2. 5 沉积锰价态
LiMn2O4对应与四种阳极材料(石墨、Li4Ti5O12、去锂LiFePO4、金属Li)组装成电池,经过100次室温循环后,四种阳极材料XANES分析结果如图11(a),分析表明在阳极上的锰价态为+2,不随充放电过程变化[26]。
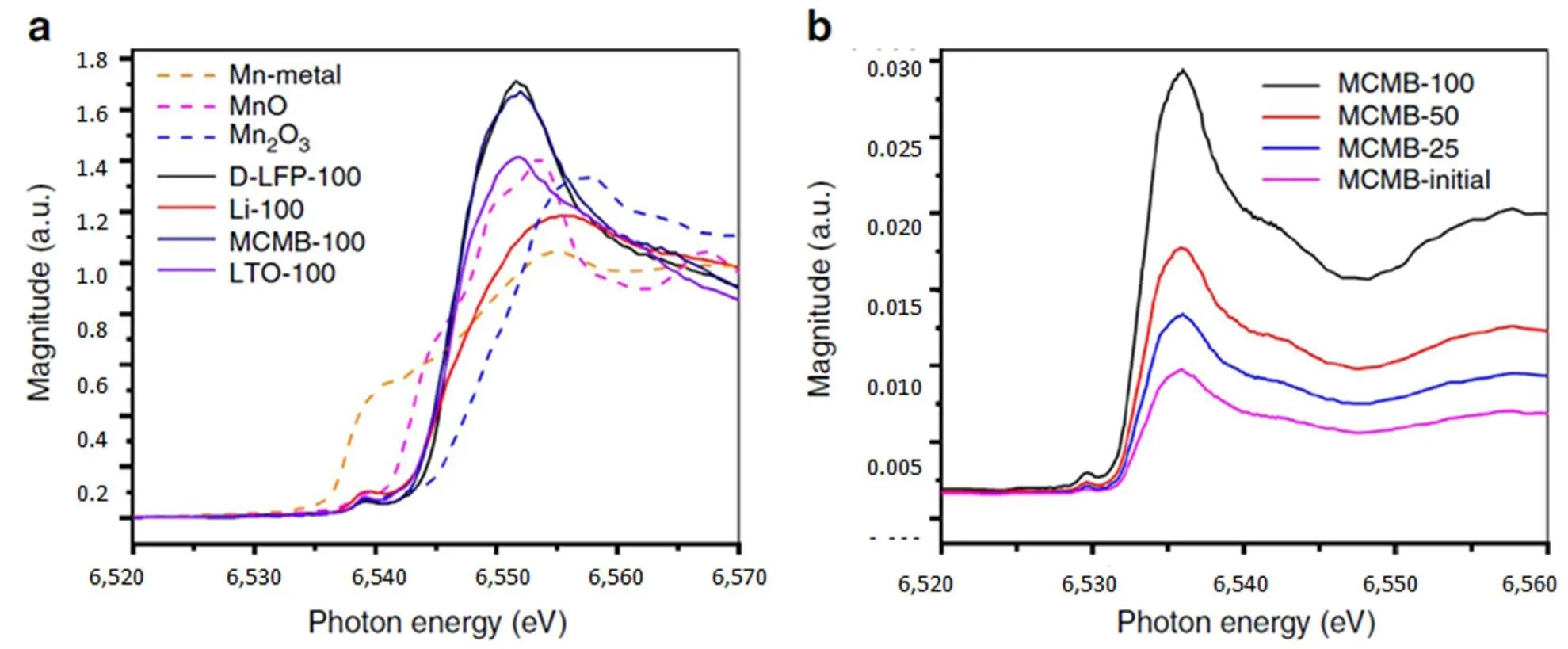
图11 X射线吸收近边谱(XANES)
图11(b)为MCMB阳极在循环前后的XANES图谱,在6,545 eV光子能量处(表明是二价锰),其荧光强度随循环次数的增加而增强,即随循环次数的增加,锰沉积量越多,与前所述一致[26]。这个实验结果有力地证实了:锰离子在阳极SEI上沉积之后,并不是如早期文献所述经过还原反应生成了金属锰,而是继续以二价态存在。
3 容量衰减新机理
3.1 实验总结
通过对以上实验数据分析,我们得出以下结论:(1) 与阳极相比,阴极衰减所引起电池体系容量衰减量可以忽略。(2) 沉积在石墨SEI膜上的锰量随着电池循环次数增加而增多,电池容量衰减得越多。(3) 由于Mn2+不断迁移到石墨阳极,改变了其SEI系能,从而导致了锰酸锂体系电池容量的衰减。(4) 阳极上锂离子越多,电池容量衰减越快,这暗示了在SEI膜中的锂离子与锰离子进行了离子交换。(5) 锂离子镶锂/脱锂过程中在SEI膜的进出通道越多,充电倍率能更高,且能维持更多电池容量。相反,循环后的MCMB微碳阳极SEI的进出通道少,电池容量就会变小,确切地说是电池充电功率变小。这也暗示了在SEI膜中的锂离子与锰离子进行了离子交换。(6) 沉积在阳极上的锰价态为+2。
3.2 推导出新观点

图12 锰沉积导致容量衰减的机理示意图
以上结论从逻辑上支持了“锰沉积导致尖晶石锰酸锂/石墨体系的锂离子电池容量衰减”新机理。锰沉积导致容量衰减的新机理示意图如图12:Mn (II)与SEI中的活性锂离子进行离子交换,沉积在阳极上,堵塞了锂离子进出通道,导致容量衰减。
为什么Li4Ti5O12阳极在工作环境在室温下、甚至在55℃时,容量衰减依然缓慢[36],但在80℃却表现出剧烈衰减[42],怎样解释这一现象?根据新机理,我们认为与石墨阳极相比,Li4Ti5O12阳极有足够多的通道,即使在55℃时堵塞了部分通道,但还有大量的通道,并不影响锂离子进出,所以容量维持率高,但当锰离子堵塞了更多的通道之后,锂离子能进出的通道不多时,阻碍锂离子进出,此时会表现出显著的容量衰减,与石墨阳极容量急剧衰减的情况相似。
[1] Armand M, Tarascon J M. Building better batteries[J]. Nature, 2008, 451(7179): 652-657.
[2] Hibino M, Nakamura M, Kamitaka Y, et al. Improvement of cycle life of spinel type of lithium manganese oxide by addition of other spinel compounds during synthesis [J]. Solid State Ionics, 2006, 177(26/32): 2653−2656.
[3] Luo J Y, Li X L, Xia Y Y. Synthesis of highly crystalline spinel LiMn2O4by a soft chemical route and its electrochemical performance [J]. Electrochimica Acta, 2007, 52(13): 4525−4531.
[4] Deng B H, Nakamura H, Yoshio M. Comparison and improvement of the high rate performance of different types of LiMn2O4spinels[J]. J Power Sources, 2005, 141(1): 116−121.
[5] Yang L, Takahashi M, Wang B. A study on capacity fading of lithium-ion battery with manganese spinel positive electrode during cycling[J]. Electrochimica Acta, 2006, 51(16): 3228-3234.
[6] Li X F, Xu Y L. Spinel LiMn2O4active material with high capacity retention[J]. Applied Surface Science, 2007, 253(21): 8592−8596.
[7] Liu D Q, Liu X Q, He Z Z. Surface modification by ZnO coating for improving the elevated temperature performance of LiMn2O4[J]. Alloys and Compounds, 2007, 436(1): 387−391.
[8] Zenghui Han,Xiaozhi Jia,Hui Zhan,et al. LiMn2O4/ LiNi0.5Mn1.5O4composite with improved electrochemical property [J]. Electrochimica Acta, 2013, (114):772-778.
[9] Ze Yang,Yan Jiang,Heng-Hui Xu,et al. High- performance porous nanoscaled LiMn2O4prepared by polymer-assisted sol–gel method [J]. Electrochimica Acta , 2013, 106 :63– 68.
[10] Zhang X, Cheng F, Yang J, et al. LiNi0.5Mn1.5O4porous nanorods as high-rate and long-life cathodes for Li-ion batteries[J]. Nano letters, 2013, 13(6): 2822-2825.
[11] Sen Zhao,Ying Bai,Qingjun Chang, et al. Surface modification of spinel LiMn2O4with FeF3 for lithium ion batteries [J]. Electrochimica Acta, 2013, (108):727-773.
[12] Cheng J H, Hy S, Rick J, et al. Mechanistic Basis of Enhanced Capacity Retention Found with Novel Sulfate-Based Additive in High-Voltage Li-Ion Batteries[J]. The Journal of Physical Chemistry C, 2013, 117(44): 22619-22626.
[13] Pieczonka N P W, Liu Z, Lu P, et al. Understanding transition-metal dissolution behavior in LiNi0. 5Mn1. 5O4High-Voltage spinel for lithium ion batteries[J]. The Journal of Physical Chemistry C, 2013, 117(31): 15947-15957.
[14] Zheng H H, Sun Q , Liu G., et al. Correlation between dissolution behavior and electrochemical cycling performance for LiNi1/3Co1/3Mn1/3O2-based cells [J]. Journal of Power Sources, 2012, (207):134–140.
[15] Zheng H H, Sun Q , Liu G., et al. Correlationship between electrode mechanics and long-term cycling performance for graphite anode in lithium[J]. Journal of Power Sources, 2012 (217): 530-537.
[16] Choa J, Thackeray M M. Structural Changes of LiMn2O4Spinel Electrodes during Electrochemical Cycling[J]. Journal of Electrochemical Society, 1999, 146(10): 3577-3581.
[17] Gnanaraj J S, Pol V G, Gedanken A, et al.. Improving the high temperature performance of LiMn2O4spinel electrodes by coating the active mass via a sonochemical method [J]. Electrochemistry Communications, 2003, 5(11): 940-945.
[18] Yu L H, Qiu X P, Xi J Y ,et al. Enhanced high potential and elevated-temperature cycling stability of LiMn2O4cathode by TiO2modification for Li-ion battery [J]. Electrochimica acta, 2006, 51(28): 6406-6411.
[19] Walz K A, Johnson C S, Genthe J, et al. Elevated temperature cycling stability and electrochemical impedance of LiMn2O4cathodes with nanoporous ZrO2and TiO2coatings [J]. Journal of power sources, 2010, 195(15): 4943-4951.
[20] Hirayama M, Ido H, Kim K S, et al. Dynamic structural changes at LiMn2O4/electrolyte interface during lithium battery reaction [J]. Journal of the American Chemical Society, 2010, 132(43): 15268-15276.
[21] Blyr, A, Sigala C, Amatucci G,et al. Self-discharge of LiMn2O4/C Li-ion cells in their discharged state [J]. J. Electrochem. Soc. 1998, 145(1):194-209.
[22] Amine K,Liu J,Kang S,et al. Improved lithium manganese oxide spinel/graphite Li-ion cells for high-power applications[J]. Journal of power sources, 2004, 129(1): 14-19.
[23] Komaba S,Kumagai N,Kataoka Y. Influence of manganese (II), cobalt (II), and nickel (II) additives in electrolyte on performance of graphite anode for lithium-ion batteries[J]. Electrochimica acta, 2002, 47(8): 1229-1239.
[24] Dai Y, Cai L, White R E. Capacity fade model for spinel liMn2O4electrode [J].Journal of The Electrochemical Society, 2013,160 (1):182-190.
[25] Lin X, Park J, Liu L, et al. A comprehensive capacity fade model and analysis for li-ion batteries[J]. Journal of The Electrochemical Society, 2013,160(10): 1701-A1710.
[26] Zhan C, Lu J, Kropf A J, et al. Mn (II) deposition on anodes and its effects on capacity fade in spinel lithium manganate–carbon systems[J]. Nature communications, 2013(10): 1-8.
[27] Thackeray M M,Wolverton C, Isaacs E. Electrical energy storage for transportation approaching the limits of, and going beyond, lithium-ion batteries [J]. Energy & Environmental Science, 2012, 5(7): 7854-7863.
[28] Vetter J,Novak P, Wagner M R, et al. Ageing mechanisms in lithium-ion batteries [J]. Journal of power sources, 2005, 147(1): 269-281.
[29] Chung K Y, Lee H S, Yoon W S, et al. Studies of LiMn2O4capacity fading at elevated temperature using in situ synchrotron X-ray diffraction[J]. Journal of The Electrochemical Society, 2006, 153(4): A774-A780.
[30] Aurbach D, Markovsky B, Salitra G., et al. Review on electrode-electrolyte solution interactions, related to cathode materials for Li-ion batteries [J]. Journal of Power Sources, 2007, 165(2): 491-499.
[31] Wohlfahrt-Mehrens M, Vogler C, Garche J. Aging mechanisms of lithium cathode materials[J]. J.Power Sources, 2004, 127(1): 58-64.
[32] Amine K,Liu J,Kang S,et al. Improved lithium manganese oxide spinel/graphite Li-ion cells for high-power applications [J]. J.Power Sources, 2004, 129(1): 14-19.
[33] Broussely M, Biensan P, Bonhomme F, et al. Main aging mechanisms in Li ion batteries[J]. Journal of power sources, 2005, 146(1-2): 90-96.
[34] Verma P, Maire P, Novák P. A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries[J].Electrochimica Acta,2010,55(22): 6332-6341.
[35] Jones J, Anouti M, Caillon-Caravannier M, et al. Solubilization of SEI lithium salts in alkylcarbonate solvents [J].Fluid Phase Equilibria,2011, 305(2):121-126.
[36] Huang C, Huang K,Wang H, et al. The effect of solid electrolyte interface formation conditions on the aging performance of Li-ion cells[J]. Journal of Solid State Electrochemistry, 2011, 15(9): 1987-1995.
[37] Kassem M, Bernard J, Revel R, et al. Calendar aging of a graphite/LiFePO4cell [J]. J. Power Sources, 2012, 208: 296-305.
[38] Delacourt C. Modeling Li-Ion batteries with electrolyte additives or contaminants[J]. Journal of The Electrochemical Society, 2013, 160(11): 1997-2004.
[39] Wu Y, Reddy M V, Chowdari B V R, et al. Long-term cycling studies on electrospun carbon nanofibers as anode material for lithium ion batteries[J]. ACS applied materials & interfaces, 2013, 5(22): 12175-12184.
[40] Komaba S, Itabashi T, Ohtsuka T, et al. Impact of 2-Vinylpyridine as electrolyte additive on surface and electrochemistry of graphite for C/LiMn2O4Li-Ion Cells [J]. Journal of The Electrochemical Society, 2005, 152(5): A937-A946.
[41] Ochida M, Domi Y, Doi T, et al. Influence of manganese dissolution on the degradation of surface films on edge plane graphite negative-electrodes in lithium-ion batteries[J]. Journal of The Electrochemical Society, 2012, 159(7): A961-A966.
[42] Amine K, Chen Z, Zhang Z, et al. Mechanism of capacity fade of MCMB/Li1.1(Ni1/3Mn1/3Co1/3)0.9O2cell at elevated temperature and additives to improve its cycle life [J]. Journal of Materials Chemistry,2011,21(44): 17754-17759.
[43] Barsoukov E, Kim J H, Kim J H, et al. Kinetics of lithium intercalation into carbon anodes: in situ impedance investigation of thickness and potential dependence[J]. Solid State Ionics, 1999, 116(3): 249-261.
[44] Peled E, Bar Tow D, Merson A, et al. Composition, depth profiles and lateral distribution of materials in the SEI built on HOPG-TOF SIMS and XPS studies[J]. Journal of power sources, 2001, 97-98: 52-57.
[45] Li J T, Chen S R, Fan X Y, et al. Studies of the interfacial properties of an electroplated Sn thin film electrode/electrolyte using in situ MFTIRS and EQCM[J]. Langmuir, 2007, 23(26): 13174-13180.
[46] Liu X H, Huang J Y. In situ TEM electrochemistry of anode materials in lithium ion batteries[J]. Energy & Environmental Science, 2011, 4(10): 3844-3860.
[47] Wang Z, Xie G, Gao L. Electrochemical characterization of Li4Ti5O12/C anode material prepared by starch-sol-assisted rheological phase method for Li-ion battery[J]. Journal of Nanomaterials, 2012, 2012: 9.
[48] Tsunekawa H, Tanimoto S, Marubayashi R, et al. Capacity fading of graphite electrodes due to the deposition of manganese ions on them in Li-ion batteries[J]. Journal of The Electrochemical Society, 2002, 149(10): A1326-A1331.
[49] Li Tan, Li Zhang, Qingna Sun, et al. Capacity loss induced by lithium deposition at graphite anode for LiFePO4/graphite cell cycling at different temperatures [J]. Electrochimica Acta , 2013,111 :802- 808.
MECHANISM ANALYSIS ON CAPACITY FADING OF SPINEL LITHIUM MANGANESE OXIDE CELL
*CHEN Hai-hui1,2, QIU Xin-ping2
(1. School of Chemistry and Chemical Engineering,Jinggangsshan University, Ji’an, Jiangxi, 343009, China;2. Department of Chemistry, Tsinghua University, Beijing, 100084, China)
The capacity fade of spinel lithium manganese oxide cell is a bottleneck challenge for its large-scale application. It is traditionally thought that Mn(II) ions at the anode is reduced to the metallic manganese that helps for catalyzing electrolyte decomposition. This could poison and damage the solid electrolyte interface (SEI) film leading to the capacity fade in Li-ion batteries. But the latest research has identified that the manganese deposited on the anode is +2 valent, and is not reduced to the metallic manganese. Research progresses of capacity fade of manganese lithium ion batteries in the world were introduced in this paper, based on the new discovery “manganese deposited on anode is +2 valent” and the results obtained from the literature, a new mechanism has been purposed that Mn(II) is exchanged with active Li-ion in anode SEI film, then deposited at the anode, which hinders and/or blocks the intercalation/de-intercalation of lithium ions, then leads to the capacity fade in Li-ion batteries.
capacity fade; manganese deposition; lithium manganese oxide; lithium-ion batteries
1674-8085(2015)04-0034-09
TM 912.1
A
10.3969/j.issn.1674-8085.2015.04.007
2015-01-06;修改日期:2015-03-07
江西省科技支撑计划项目(20122BBE500047、20123BBG70204);江西省教育厅科技计划项目(KJLD12080,2013年度访学项目)
*陈海辉(1968-),男,湖南衡山人,副教授,博士,主要从事化学工程与工艺研究(E-mail:chenhaihui68@126.com);
邱新平(1966-),男,江苏南京人,教授,博士,主要从事化学新能源研究(E-mail:qiuxp@mail.tsinghua.edu.cn).
