流感病毒两种反向遗传学操作系统的初步构建
2015-11-20连孟洋武伟华董方圆丁小满房师松
连孟洋,王 昕,彭 博,武伟华,刘 慧,董方圆,丁小满,房师松,郑 青
2.深圳市疾病预防控制中心,深圳 518055;
3.中山大学公共卫生学院,广州 510080
当今世界处于后流感大流行期,甲型流感病毒在大范围内季节性传播,并感染包括人、禽类等多个物种,故世界各地都积极致力对此病毒的研究。甲型流感病毒是单股负链分节段的RNA病毒,病毒基因组约13.6kb,分为8个大小不等的独立片段,携带至少13种病毒蛋白[1]。流感病毒基因组RNA含多个开放阅读框,并与3′和5′端非编码区相连[2]。近年来,负链RNA病毒的基因操作基于反向遗传学技术获得突破性进展,同时流感病毒反向遗传学研究也在其生命周期等方面取得很大进展[3]。就甲型流感病毒而言,Luytjes和 Enami等[4-5]建立基于体外重组核糖核蛋白复合体(vRNPs)转染辅助病毒感染性细胞的反向遗传学系统,vRNPs是由体外转录产生的RNA及纯化的病毒核蛋白(NP)和聚合酶蛋白(PB1,PB2和PA)孵育而成。辅助病毒则可作为病毒核蛋白和聚合酶蛋白的来源以帮助vRNPs在细胞内复制。随后,Zobel等[6]和 Neumann等[7]提出依赖 RNA 聚合酶I的体内合成vRNA的反向遗传学系统,成功获得流感病毒cDNA 的精确复制[7]。1996年,Pleschka等[8]提出不依赖辅助病毒的反向遗传学系统—利用人RNA聚合酶I启动子及丁型肝炎病毒基因组核糖酶来完成反向遗传操作。同年,Lutz等[9]得出甲型流感病毒表达报告基因可作为检测和定量病毒复制的工具。随后,Neumann等[10]同样不依赖辅助病毒,利用12个质粒共转染293T细胞,在人RNA聚合酶Ⅰ启动子、小鼠RNA聚合酶Ⅰ终止子以及RNA聚合酶Ⅱ的调控下,成功包装 A/WSN/33(H1N1)病 毒 粒 子。Hoffmann 等[11]在 Neumann[10],Fodor[12]等研究成果的基础上于2000年建立了基于8质粒的双向RNA聚合酶Ⅰ/聚合酶Ⅱ转录系统,此系统允许甲型流感病毒基于8个质粒拯救其子代病毒,使所需质粒数量由12/17降低到8,减少其所需时间及成本,促进双向的反向遗传学系统的发展。
本研究基于甲型流感病毒H1N1分离株A/Puerto Rico/8/34的基因组,分别构建依赖辅助病毒和宿主细胞内合成vRNPs的反向遗传学操作系统,并尝试利用上述两系统表达EGFP,为本课题后期反向遗传学系统的建立提供借鉴。
1 材料与方法
1.1 材料
1.1.1 病毒毒株 人源流感病毒 A/Puerto Rico/8/34(H1N1),由深圳市疾病预防与控制中心提供。
1.1.2 载体 克隆载体 pBluescript II sk(+)(Takara公司),pcDNA3.1(+)(Invitrogen公司)。
1.1.3 大肠杆菌菌种和细胞系 大肠杆菌JM109感受态细胞 (Takara公司);MDCK细胞,293T细胞,由深圳市疾病预防与控制中心提供。
1.1.4 引物
1.1.4.1 构建POL I系统(pBluescript II sk(+)-tI-EGFP-PIh),设计引物见表1。
1.1.4.2 构建表达载体 pcDNA3.1-PB2 、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP 设 计引物见表2。

表1 构建pBluescript II sk(+)-tI-EGFP-PIh载体的引物Tab.1 Primers for pBluescript II sk(+)-tI-EGFP-PIh
上述引物均合成于Takara公司。
1.1.5 主要试剂 PCR扩增试剂盒(Takara公司),质粒DNA抽提纯化试剂盒(Takara公司),T4 DNA连接酶(Takara公司),PCR产物回收试剂盒(QIAGEN公司),胶回收试剂盒(QIAGEN公司),无内毒素的质粒DNA抽提纯化试剂盒(QIAGEN公司),限制性内切酶 HindIII、XhoI、BamhI、NotI、XbaI(NEB 公 司 ),转 染 试 剂 盒 Lipofectamine2000(Invitrogen 公 司 ),细 胞 培 养 基DMEM(Gibco公司)、Opti-MEM(Gibco公司),胎牛血清FBS(Gibco公司),双抗青霉素、链霉素(Invitrogen公司),EDTA 胰酶(Gibco公司),TPCK处理胰酶(Gibco公司)。

表2 构建pcDNA3.1-PB2、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP载体的引物Tab.2 Primers for pcDNA3.1-PB2 、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP
1.2 方法
1.2.1 载体的构建
1.2.1.1 POL I系统(pBluescript II sk(+)-tI-EGFP-PIh)的构建 经文献所述,因鼠源聚合酶I终止子序列末端6个腺嘌呤(A)碱基的差异[13],在此设计两个POL I系统(POL I系统1和POL I系统2)。
为了构建POL I系统,首先利用聚合酶I元件引物扩增聚合酶I元件1和2序列,然后分别使用相应的引物将人POL I启动子PIh、鼠的终止子tI及EGFP基因克隆出,随后基于嵌合引物合成tI-EGFP-PIh基因片段,最后经限制性内切酶Hind III和Xho I酶切,T4DNA连接酶连接,将其克隆至pBluescript II sk(+)载体。
1.2.1.2 表达载体pcDNA3.1-PB2 、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP的构建 PB2、PB1、PA、NP基因的cDNA和载体pcDNA3.1经限制性内切酶酶切,连接获得重组质粒pcDNA3.1-PA 、pcDNA3.1-PB1、pcDNA3.1-PB2、pcDNA3.1-NP。
上述所有重组质粒均经过菌液PCR、双酶切鉴定和测序分析以保证载体的正确。
1.2.2 细胞转染
1.2.2.1 MDCK细胞转染 MDCK细胞在25m2细胞培养瓶中长至90%左右,用预温的PBS液洗3次,EDTA胰酶消化细胞并按照1∶10的比例铺6孔细胞培养板。待细胞密度为80%~90%时,用PBS液洗3次,将培养基换为病毒接种液,随后用10μL流感病毒H1N1毒株(血凝价为1∶4)感染细胞。感染2h后,用PBS液洗3次,按照Lipofectamine 2000说明书进行操作,将纯化质粒pBluescript II sk(+)-tI-EGFP-PIh-1和 pBluescript II sk(+)-tI-EGFP-PIh-2分别转染至 MDCK细胞中。
1.2.2.2 293T细胞转染 293T细胞在25m2细胞培养瓶中长至90%左右,用PBS液洗3次,按照1∶6的比例铺6孔细胞培养板。待细胞密度为80-85%时,用 PBS液 洗3次,按 照 Lipofectamine2000说明书进行操作,将纯化质粒pcDNA3.1-PB2 、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP 与 pBluescript II sk(+)-tI-EGFPPIh-1/2分别共转染293T细胞。
2 结 果
2.1 POL I系统(pBluescript II sk(+)-tI-EGFPPIh)的构建 研究构建的 pBluescript II sk(+)-tI-EGFP-PIh载体的示意图,见图1。

图1 pBluescript II sk(+)-tI-EGFP-PIh载体的示意图Fig.1 Schematic diagram of pBluescript II sk(+)-tI-EGFPPIh.
2.2 表达载体pcDNA3.1-PB2 、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP的构建,见图2。
2.3 细胞转染
2.3.1 依赖于辅助病毒的转染,见图3。
转染时,需先用流感病毒 H1N1(血凝价为1∶4)感染MDCK细胞2h,随后将含有POL I系统的 pBluescript II sk(+)-tI-EGFP-PIh-1/2 重组质粒转染MDCK细胞,同时,分别将pEGFP-N1和pBluescript II sk(+)质粒转染MDCK细胞作为阳性和阴性对照。转染后12h,用荧光显微镜观察到细胞开始出现荧光,在转染48h时,荧光最强。同时,分别经 pBluescript II sk(+)-tI-EGFP-PIh-1和pBluescript II sk(+)-tI-EGFP-PIh-2转染的两组细胞,其荧光强度没有明显差别,见图4。
图2 pcDNA3.1-PB2 、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP表达载体的示意图Fig.2 Schematic diagram of expression vectors--- pcDNA3.1-PB2、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP.
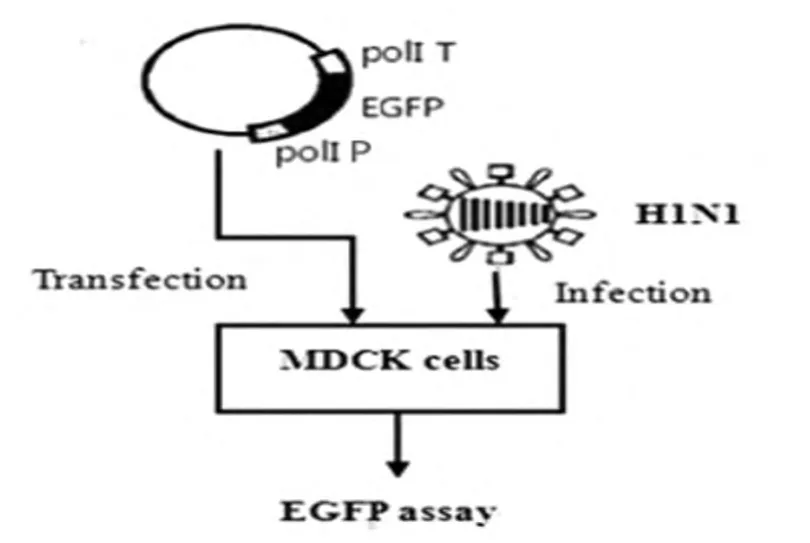
图3 依赖于辅助病毒的POL I系统转染示意图Fig.3 Schematic diagram of transfection that POLI system relying on helper virus.
2.3.2 依赖宿主细胞内合成RNPs的转染,见图5。
将表达载体pcDNA3.1-PB2、pcDNA3.1-PB1、pcDNA3.1-PA、pcDNA3.1-NP与 POL I系统质粒pBluescript II sk(+ )-tI-EGFP-PIh-1/2 共 转 染293T细胞,同时,分别设pEGFP-N1转染阳性对照组和pBluescript II sk(+)转染阴性对照组。转染12h后,在荧光显微镜下可观察到荧光现象,细胞培养至48h时,荧光强度逐渐增强。与上组依赖辅助病毒的转染实验相同,分别经POL I系统pBluescript II sk(+)-tI-EGFP-PIh-1和 pBluescript II sk(+)-tI-EGFP-PIh-2 转 染 的 两 组 细 胞,其 荧 光 强 度均较低,见图6。
上述实验结果表明:依赖辅助病毒和依赖宿主细胞内合成RNPs的转染系统均可使外源基因EGFP在哺乳动物细胞中表达。
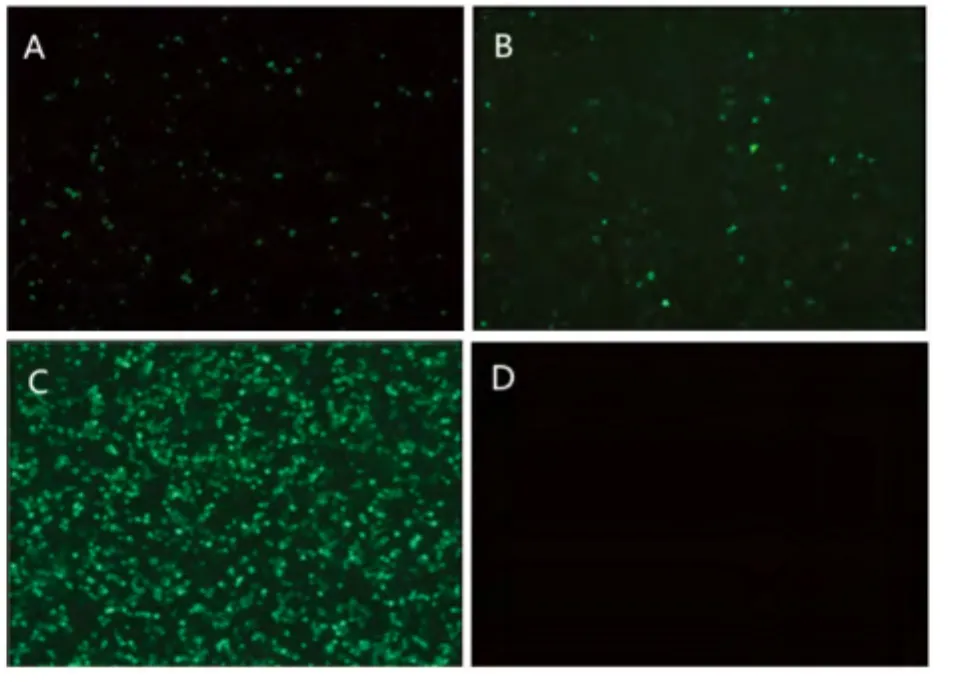
图4 依赖辅助病毒的POL I系统转染MDCK细胞48h的绿色荧光蛋白表达Fig.4 The expression of Green Fluorescent protein at 48h post-transfection that POLI system based upon helper virus-infection.
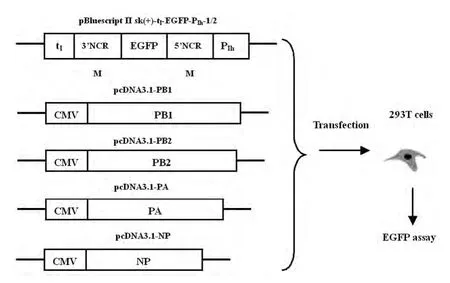
图5 依赖宿主细胞内合成vRNPs的转染Fig.5 The transfection of virus ribonucleoprotein complex(vRNPs)that synthetised in the host cell.
3 讨 论
流感病毒等负链RNA病毒因其RNA为负极性,故其vRNA对细胞不具感染性。对流感病毒而光强度较弱,其原因可能与转染效率有关,因五个质粒共转染293T细胞需要五个质粒进入同一个细胞,此项工作较为困难;并且五质粒转染时所加的转染试剂对细胞有很大伤害;同时与宿主细胞内vRNPs表达效率有关。综上所述,依赖辅助病毒或vRNPs的反向遗传学操作系统均可驱动外源蛋白的表达,促进我们更进一步来研究流感病毒的反向遗传学。言,最小病毒复制单位为vRNPs,它由聚合酶蛋白(PB2,PB1,PA)、核蛋白(NP)及vRNA 组成。此vRNPs既可以在细胞外[4],亦可在细胞内合成[7],但最终都进入细胞核,使得流感病毒RNA经复制、转录为 mRNA、cRNA 及负链vRNA。因此,vRNPs可作为病毒复制的前提,使得病毒蛋白在其基础上表达。
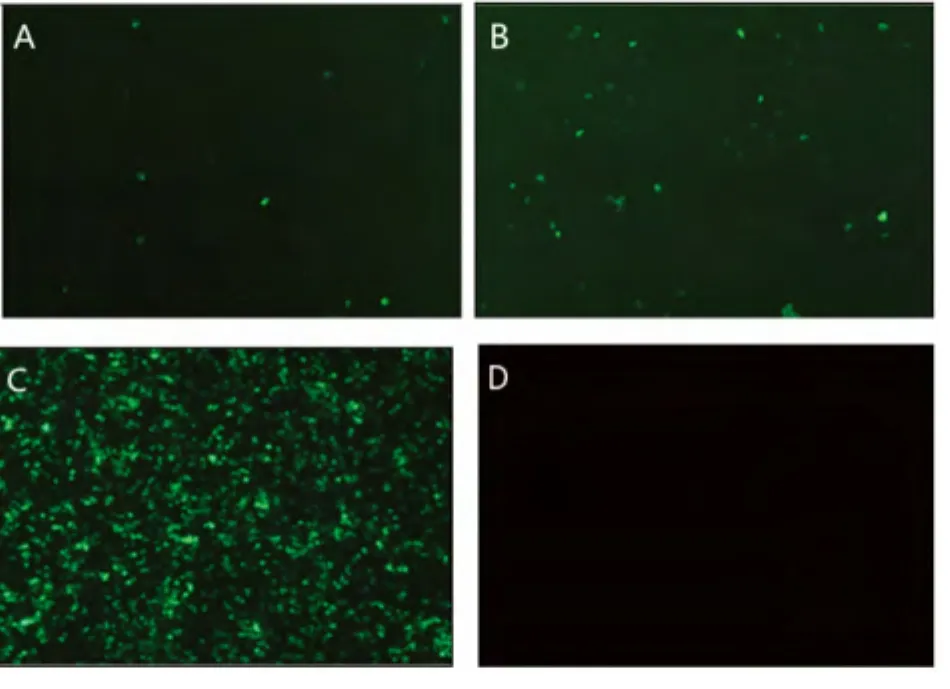
图6 vRNPs系统转染293T细胞48小时的绿色荧光蛋白表达Fig.6 The expression of Green Fluorescent protein at 48h post-transfection that vRNPs system.
本研究构建了依赖辅助病毒和宿主细胞内合成核糖核蛋白复合体两个反向遗传学操作系统,并在其基础上表达外源绿色荧光蛋白。在实验中,两个反向遗传学操作系统均可使绿色荧光蛋白表达,在荧光显微镜下均可观察到荧光的发生,由此说明:本实验所构建的聚合酶I系统在驱动外源蛋白在细胞内表达上是有效的。从图片显示,两个转染系统的荧光强度不高。由于POLI系统中的启动子为人RNA聚合酶I启动子,且RNA聚合酶I转录系统具有种属特异性,但基于高等哺乳动物RNA POLI启动子亦可推动低等哺乳动物RNA转录启动的理论[14],本研究中两个反向遗传学系统均可驱动EGFP转录的启动。同时,本实验所构建的POLI系统中的启动子序列前加有增强子,因此在宿主细胞中POLI系统的转录效率应较高,对荧光强度的影响较小。在依赖辅助病毒的转染系统中,荧光强度不高的原因主要有:脂质体2000对MDCK细胞的转染效率本就不高,转染试剂亦对细胞有损伤;此外,因辅助病毒滴度较低,其对MDCK的感染效率也是一个因素。在细胞内合成vRNPs的转染体系中,荧
[1]Hsu JT,Yeh JY,Lin TJ,et al.Identification of BPR3P0128as an inhibitor of cap-snatching activities of influenza virus[J].Antimicrob Agents Chemother,2012,56(2):647-657.
[2]Desselberger U,Racaniello V R,Zazra J J,and Palese P.The3′and 5′-terminal sequences of influenza A,B and C virus RNA segments are highly conserved and show partial inverted complementarity[J].Gene,8,1980,8(3):315-328.
[3]Wit E,Spronken MJ,Bestebroer TM,et al.Efficient generation and growth of influenza virus A/PR/8/34from eight cDNA fragments[J].Virus Res,2004,103(1-2):155-161.
[4]Luytjes W,Krystal M,Enami M,et al.Amplification,expression,and packaging of a foreign gene by influenza virus[J].Cell,1989,59(6):1107-1113.
[5]Enami M,Luytjes W,Krystal M,et al.Introduction of site-specific mutations into the genome of influenza virus[J].Proc Natl Acad Sci USA ,1990(10):3802-3805.
[6]Zobel A,Neumann G,Hobom G.RNA polymerase I catalysed transcription of insert viral cDNA[J].Nucleic Acids Res,1993,21(16):3607-3614.
[7]Neumann G,Zobel A,Hobom G.RNA polymerase immediated expression of influenza viral RNA molecules[J].Virol,1994,202(1):477-479.
[8]Pleschka S,Jaskunas SR,Engelhardt OG,et al.A plasmidbased reverse genetics system of influenza A virus[J].J Virol,1996(70):4188-4192.
[10]Lutz A,Dyall J,Olivo PD,Pekosz A.Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication[J].Journal of virological methods,2005,126(1-2):13-20.
[10]Neumann G,Watanabe T,Ito H,et al.Generation of influenza A viruses entirely from cloned cDNAs[J].Proc Natl A cad Sci,1999(96):9345-9350.
[11]Hoffmann E,Neumann G,Hobom G,et al."Ambisense"approach for the generation of influenza a virus:vRNA and mRNA synthesis from one template[J].Virol,2000,267(2):310-317.
[12]Fodor E,Devenish L,Engelhardt OG,et al.Rescue of influenza A virus from recombinant DNA[J].J Virol,1999(73):9679-9682.
[13]Zobel A,Neumann G,Hobom G.RNA polymerase I catalysed transcription of insert viral cDNA[J].Nucleic Acids Research,1993,21(16):3607-3614.
[14]Heix J,Grummt I.Species specificity of transcription by RNA polymerase I[J].Current Opinion in Genetics & Development,1995,5(5):652-656.
