添加扩增内标的PCR方法快速检测食品中的沙门氏菌
2015-11-07张德福付绪磊汤轶伟白凤翎杨文慧赵丽红张义全励建荣渤海大学食品科学与工程学院辽宁省食品安全重点实验室生鲜农产品贮藏加工及安全控制技术国家地方联合工程研究中心辽宁锦州0军事医学科学院微生物流行病研究所病原微生物生物安全国家重点实验室北京0007江苏大学医学院江苏镇江0渤海大学数理学院辽宁锦州0
张德福,付绪磊,汤轶伟,白凤翎,殷 喆,杨文慧,赵丽红,张义全,李 春,励建荣,*(.渤海大学食品科学与工程学院,辽宁省食品安全重点实验室,生鲜农产品贮藏加工及安全控制技术国家地方联合工程研究中心,辽宁锦州0;.军事医学科学院微生物流行病研究所,病原微生物生物安全国家重点实验室,北京0007;.江苏大学医学院,江苏镇江0;.渤海大学数理学院,辽宁锦州0)
添加扩增内标的PCR方法快速检测食品中的沙门氏菌
张德福1,2,付绪磊1,汤轶伟1,白凤翎1,殷喆2,杨文慧2,赵丽红1,张义全3,李春4,励建荣1,*
(1.渤海大学食品科学与工程学院,辽宁省食品安全重点实验室,生鲜农产品贮藏加工及安全控制技术国家地方联合工程研究中心,辽宁锦州121013;2.军事医学科学院微生物流行病研究所,病原微生物生物安全国家重点实验室,北京100071;3.江苏大学医学院,江苏镇江212013;4.渤海大学数理学院,辽宁锦州121013)
为了建立一种含扩增内标的沙门氏菌PCR检测方法,以细菌16S rRNA为扩增内标对照,以沙门氏菌invA基因为靶基因设计了一对引物,并优化了PCR反应体系。通过对20种细菌进行PCR检测显示,该方法对沙门氏菌具有良好的特异性。灵敏度实验表明,该检测方法对沙门氏菌纯DNA模板的检测灵敏度为61.1 fg/μL,对沙门氏菌纯培养物的检测灵敏度为2×102cfu/mL。对人工污染蛋清的检测实验显示,沙门氏菌接种量为2 cfu/25 mL的鸡蛋清样品经过8 h增菌培养后,可被该方法检出。结果表明,该检测方法特异性强、灵敏度高,能排除沙门氏菌PCR检测方法中可能出现的假阴性现象,适用于鸡蛋等食品中沙门氏菌的快速检测。
沙门氏菌,PCR检测,扩增内标对照,假阴性
沙门氏菌(Salmonlla)是目前世界上最重要的食源性致病菌之一,食物传播被认为是人类感染沙门氏菌的主要途径,肉、蛋、奶等动物源性食品是最容易被沙门氏菌污染的食品[1]。人类食用被沙门氏菌污染的食品会引发恶心、呕吐、腹痛、腹泻、痉挛、发烧及头痛等症状[2],严重时可导致脱水死亡[3]。在世界范围内,沙门氏菌引起的中毒事件曾占全部食品中毒事件的第一位或第二位[4]。虽然目前世界上沙门氏菌病总的发病趋势有所下降,但几乎每年都有不同程度的沙门氏菌中毒事件的报道[5]。为有效的预防和控制沙门氏菌疾病发生,建立快速有效的沙门氏菌检测方法是非常必要的。目前,法定的沙门氏菌检测方法仍采用传统培养法,操作繁琐,耗时较长,且肠杆菌科的细菌间生化反应多有交叉[6]。主流的LAMP检测方法敏感性较高,极易污染造成假阳性,且引物设计要求较高。不能满足现代社会对沙门氏菌快速检测的需求。基于聚合酶链式反应(PCR)的检测技术,因其快速、灵敏、特异性强等优点,在病原微生物检测中具有巨大的优势,已广泛应用于病原微生物的检测。
近年来有报道指出,在PCR实际检测过程中,由于环境和中间处理环节带来的一些潜在PCR反应抑制剂影响PCR反应,其结果会出现假阴性现象[7]。虽然研究人员对PCR技术进行了不断的研究和改进,但仍未有消除抑制因素的有效方法,这限制了PCR技术在病原微生物检测中的应用。有研究指出,在PCR反应体系中添加指示假阴性的扩增内标对照(internal amplification control,IAC)可以有效的提高PCR检测结果的准确性,减少假阴性的扩增结果[8-9]。PCR反应体系中采用16S rRNA的通用引物27F和1492R作为扩增内标引物,只要被检测样品中含有任意细菌,即可在电泳结果中观察到一条大小为1465 bp的电泳条带,所以整个PCR反应体系只需要加入扩增内标引物27F和1492R,不需要添加扩增内标片段,就可以达到指示假阴性结果的目的。在反应体系中,若食品样品未受沙门氏菌污染,应有扩增内标片段的扩增产物。而当PCR反应没有产生任何扩增产物时,说明反应可能受到了抑制,产生了假阴性,需重新处理待检食品样品。
invA基因是位于沙门氏菌毒力岛1上的一段高度保守序列[10],几乎所有血清型的沙门氏菌均含有invA基因[11],目前invA基因已成为沙门氏菌检测的重要靶向基因[12]。本研究以细菌16S rRNA为扩增内标对照,以编码沙门氏菌侵袭蛋白的invA基因为靶基因设计引物,建立了快速检测鸡蛋及蛋制品中沙门氏菌的快速PCR检测方法,并从特异性、纯DNA和纯培养物灵敏度以及人工污染鸡蛋清样品三个方面对该检测方法进行评价。
1 材料与方法
1.1材料与仪器
实验用的20株菌株来源见表1;Taq-PCR Master Mix、细菌基因组DNA提取试剂盒、DNA Marker D2000生工生物工程(上海)股份有限公司;琼脂糖Sigma公司;LB培养基北京奥博星生物技术有限公司。
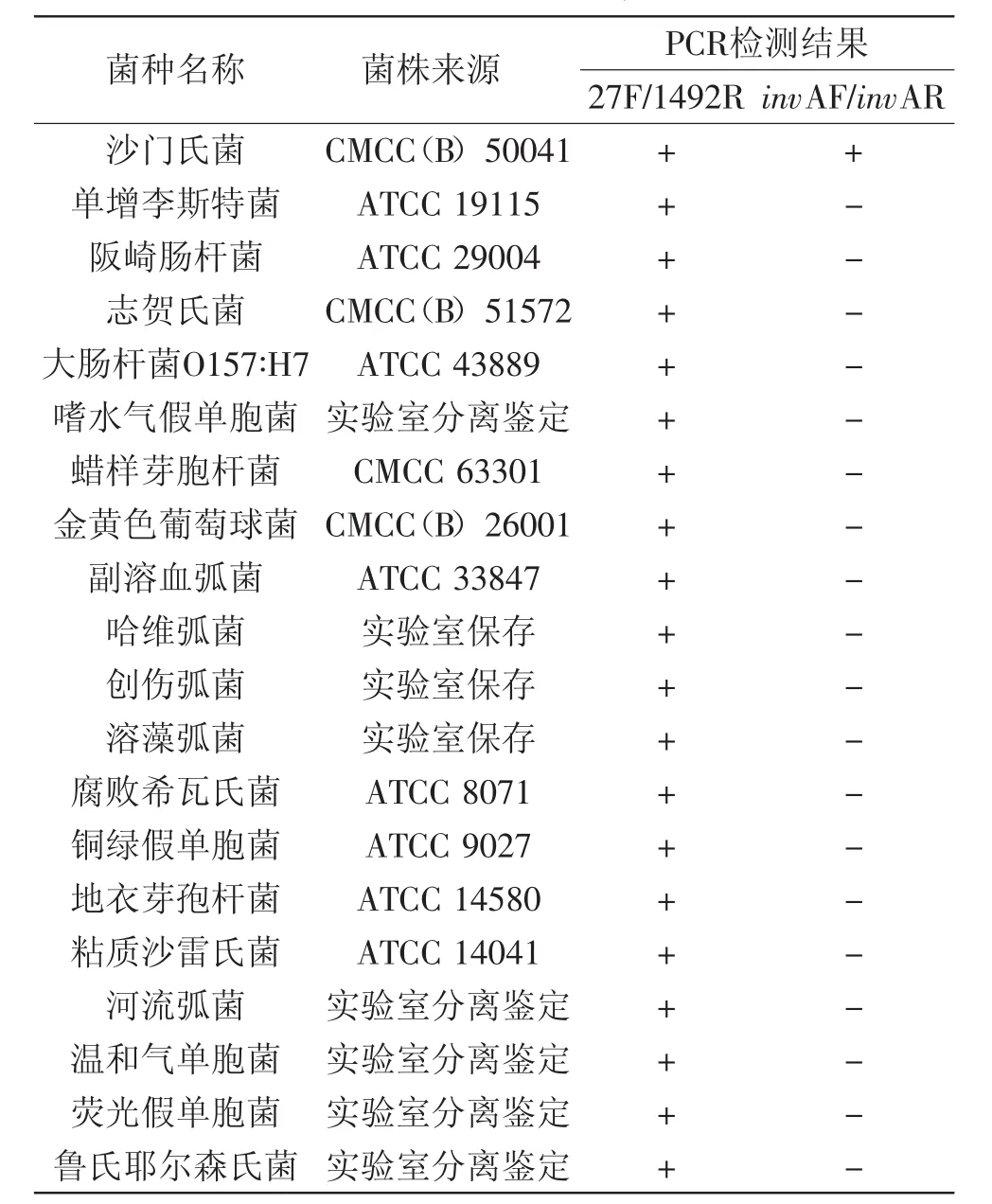
表1 实验用菌株及其PCR结果Table 1 Bacteria strains used in this study and the results of PCR
Mastercycler pro梯度PCR仪、5424R冷冻离心机德国艾本德股份有限公司;DYY-8C型电泳仪北京市六一仪器厂;Cheimd Doc XRS型凝胶成像仪美国Bio-Rad公司;NanoDrop 2000超微量分光光度计美国赛默飞世尔科技公司;DL-CJ-2N型超级洁净工作台东联哈尔(北京)仪器制造有限公司;LRH系列生化培养箱上海一恒科学仪器有限公司;LC-08拍击式均质器宁波立成仪器有限公司。
1.2实验方法
1.2.1沙门氏菌培养及DNA提取将实验室保存的菌种于LB平板上划线,37℃培养过夜。挑单菌落于LB液体培养基内,37℃培养10 h。采用细菌基因组DNA提取试剂盒提取沙门氏菌的基因组DNA,提取的DNA溶液置于-20℃冰箱保存。
1.2.2沙门氏菌基因组DNA浓度使用NanoDrop 2000超微量分光光度计测定提取的沙门氏菌基因组DNA浓度。
1.2.3引物设计和筛选选择沙门氏菌特异性基因invA(GenBank:NC_003197.1),利用软件Primer 5.0设计引物,经PCR反应验证筛选出一对特异性良好的沙门氏菌检测引物:invAF/invAR。采用细菌16S rRNA扩增通用引物27F和1492R作为PCR反应内标对照引物。实验所用引物序列如表2所示。
1.2.4PCR检测方法的建立根据Taq PCR MasterMix说明书所建议的PCR反应体系对PCR反应体系的各组分含量以及扩增程序进行微调,最终确定PCR反应最佳条件为:总体系25 μL,其中Taq PCR Master Mix 12.5 μL,DNA模板1 μL,16S rRNA引物和沙门氏菌引物终浓度均为0.4 μmol/L,补充无菌水至25 μL。以无菌水作为空白对照。

表2 引物序列Table 2 Primer sequence
PCR反应程序:94℃预变性4 min,35个循环,每个循环94℃变性30 s、53.3℃退火30 s、72℃延伸90 s,最后72℃延伸10 min。PCR产物使用1.5%的琼脂糖凝胶进行电泳,凝胶成像仪下观察结果。
1.2.5特异性验证使用细菌基因组DNA提取试剂盒提取表1中细菌的基因组DNA,并用建立的含扩增内标的PCR检测方法对所有细菌的基因组DNA进行检测,验证该PCR检测方法的特异性。
1.2.6灵敏度评价
1.2.6.1纯DNA灵敏度评价将提取的沙门氏菌基因组DNA用无菌超纯水进行10倍梯度稀释,取浓度在6.11×108~6.11 fg/μL之间的DNA溶液进行PCR扩增,凝胶成像后能得到肉眼可见条带的DNA溶液浓度即为该PCR反应的DNA灵敏度。
1.2.6.2纯培养物灵敏度评价将活化的沙门氏菌接种于LB液体培养基中,37℃培养10 h后,用无菌生理盐水进行10倍梯度稀释,取10-5、10-6、10-7、10-8四个稀释度的菌悬液进行平板计数。沙门氏菌浓度测定后,用无菌超纯水进行10倍梯度稀释,每个稀释度取1 mL置于1.5 mL离心管中。参照刘斌等[10]的方法提取沙门氏菌的基因组DNA。离心管中的菌液12000 r/min离心5 min,吸弃上清液,收集沙门氏菌菌体。用100 μL无菌超纯水重新悬浮菌体,在沸水浴中煮10 min。从沸水浴中取出后,立即在-20℃放置30 min。在37℃解冻后4000 r/min离心5 min,所得上清液作为模板进行PCR扩增,检测沙门氏菌在纯培养状态下的灵敏度。
1.2.7人工污染食品样品实验取10份鸡蛋清,每份25 mL,在超净工作台中紫外杀菌30 min。将浓度为2×109CFU/mL的沙门氏菌菌液1 mL接种于无菌蛋清中,使蛋清中沙门氏菌接种量分别为2、20、200 cfu/25 mL,每组做3个平行对照。另取1 mL 0.85%的无菌生理盐水加入无菌蛋清中做为空白对照,即接菌量为0。将上述10份人工污染样品添加到225 mL LB培养基中,37℃,150 r/min的摇床中增菌培养,每隔2 h取1 mL菌液放入1.5 mL离心管中。收集的菌液12000 r/min离心5 min,吸弃上清液,再用无菌超纯水重新悬浮菌体。离心洗涤3次后采用1.2.6.2中的实验方法提取沙门氏菌的基因组DNA作为模板进行PCR扩增。
1.2.8食品样品检测采集新鲜肉、鸡蛋、牛奶三种食品样品,共50份,每份样品取25 g或25 mL。鲜肉样品加入到盛有225 mL LB培养基的无菌均质袋中,使用拍击式均质器拍打2 min,鸡蛋及牛奶样品不进行均质,直接与培养基混匀。无菌操作将样品转移至锥形瓶中,37℃,150 r/min的摇床中增菌培养8 h。使用1.2.6.2中的方法提取基因组DNA,并用建立的PCR检测方法进行检测。
2 结果与分析
2.1基因组DNA浓度
提取的沙门氏菌基因组DNA溶液,经超微量分光光度计测定浓度为611.12 ng/μL。
2.2特异性验证
PCR检验结果如表1所示,只有沙门氏菌基因组DNA能全部扩增出大小为435 bp的检测条带和大小为1465 bp的扩增内标对照条带,而它细菌基因组DNA只能扩增出大小为1465 bp的扩增内标对照条带。表明实验所设计的引物invAF/invAR的特异性良好。
2.3灵敏度评价
2.3.1纯DNA灵敏度实验如图1所示,沙门氏菌基因组DNA经10倍梯度稀释至61.1 fg/μL时,PCR检测结果仍有肉眼可见条带,可视为沙门氏菌基因组DNA的检测限为61.1 fg/μL。

图1 沙门氏菌基因组DNA检测灵敏度Fig.1 Sensitivity of detection for Salmonella genomic DNA
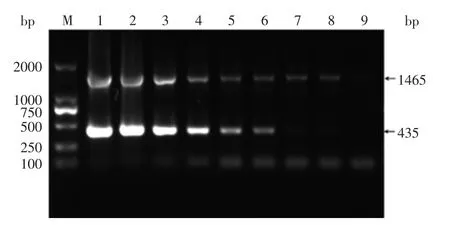
图2 沙门氏菌纯培养物检测灵敏度Fig.2 Sensitivity of detection for Salmonella pure cultures
2.3.2纯培养物灵敏度实验对培养好的沙门氏菌菌液进行平板菌落计数,沙门氏菌液浓度为2×109CFU/mL。如图2所示,沙门氏菌菌液稀释到2× 102CFU/mL时,PCR检测结果仍有肉眼可见条带,可视为沙门氏菌纯培养物检测限为2×102CFU/mL。
2.4人工污染食品样品实验
蛋清样品中接种的三个浓度的菌量分别为200、20、2 cfu/25 mL。PCR检测结果如表3所示,接菌量为200 cfu/25 mL的蛋清样品,在增菌培养6 h后即可检出。接菌量为2 cfu/25 mL和20 cfu/25 mL的蛋清样品,在增菌培养8 h后可以检出。这表明,该PCR检测方法最多经8 h增菌培养后,即可鉴定食品样品是否被沙门氏菌污染。
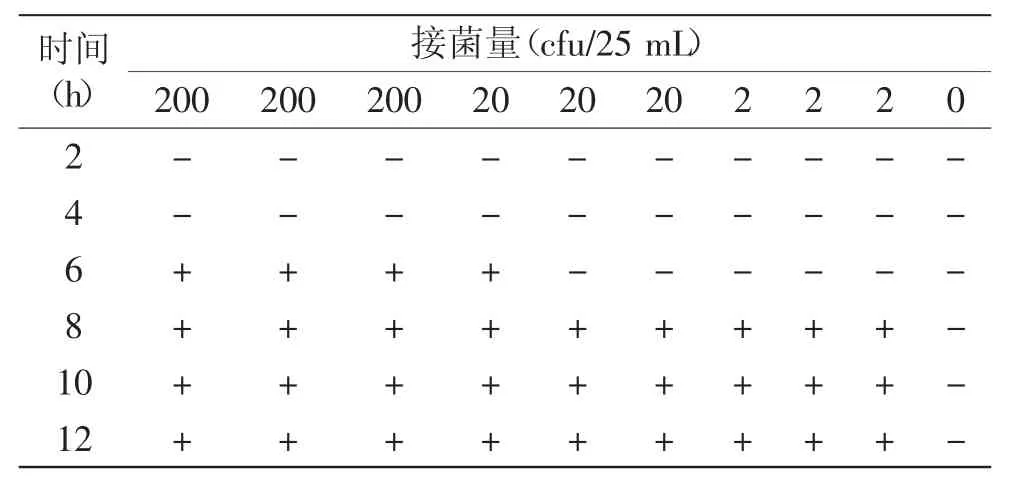
表3 人工污染食品样品检测结果Table 3 Artificially contaminated food sample test results
2.5食品样品检测结果
50份食品检测结果如表4所示,总计50份样品中19份扩增出目的条带和扩增内标条带,表明食品样品受沙门氏菌污染;28份样品中只扩增出扩增内标条带,无目的条带,表明食品未受沙门氏菌污染,3份样品未扩增出任何条带,表明样品检测过程中PCR反应受到抑制。对3份未扩增出条带的样品培养液进行稀释处理,重新检测结果显示3份样品均仅能扩增出一条内标条带。结果显示,实验采集的50份食品样品中19份受到沙门氏菌污染。

表4 食品样品检测结果Table 4 Food sample test results
3 结论
实验结果表明,以细菌16S rRNA为扩增内标对照,以沙门氏菌invA基因为检测基因建立的PCR反应体系特异性强,灵敏度高。在扩增内标存在的条件下,从沙门氏菌菌体中提取的纯基因组DNA检测灵敏度为61.1 fg/μL,沙门氏菌纯培养物检测灵敏度可达2×102CFU/mL,与刘斌、杨晋、Singh等的报道大致相同[3,13-14]。实验过程中,采用简单的煮沸裂解法代替繁琐的传统DNA提取方法提取样品中沙门氏菌的基因组DNA,既能确保低丰度的微生物被检测到,又具有步骤少、耗时短、成本低等优点[15]。沙门氏菌接种量为200、20、2 cfu/25 mL的人工污染蛋清样品,经6 h和8 h增菌培养后即可检出。这表明,在实际样品检测过程中,一般增菌6~8 h,就可满足该PCR检测方法的检出限,加上DNA模板提取、PCR扩增和电泳分析过程,整个检测至多需要12 h,远低于传统培养检测方法的4~5 d。
本研究建立的含扩增内标的沙门氏菌PCR检测方法,检测时间短,特异性好,灵敏度高,可有效排除检测过程中存在的假阴性现象。因此,该检测方法在检测沙门氏菌方面具有良好的性能,可以满足沙门氏菌快速检测的需求。
[1]Hassena A B,Barkallah M,Fendri I,et al.Real time PCR gene profiling and detection of Salmonella using a novel target:The siiA gene[J].Journal of Microbiological Methods,2015,109(1):9-15.
[2]黄文宇,柳陈坚.食源性沙门氏菌检测方法的研究进展[J].生物技术,2009,19(3):95-98.
[3]杨晋,曾庆梅,张笛,等.添加扩增内标的沙门氏菌PCR检测方法[J].生物技术通报,2014,30(7):54-59.
[4]Manzano M,Cocolin L,Astori G,et al.Development of a PCR microplate-capture hybridization method for simple,fast and sensitive detection of Salmonella serovars in food[J].Molecular and Cellular Probes,1998,12(4):227-234.
[5]王军,郑增忍,王晶钰.动物源性食品中沙门氏菌的风险评估[J].中国动物检疫,2007,24(4):23-25.
[6]陈金顶,索青利,廖明,等.沙门氏菌的invA基因序列分析与分子检测[J].中国人兽共患病杂志,2004,20(10):868-871.
[7]Malorny R,Tassios P T,Radstrom P,et al.Standardization of diagnostic PCR for the detection of foodborne pathogens[J]. International Journal of Food Microbiology,2003,83(1):39-48.
[8]Zhang W J,Cai Q,Guan X,et al.Detection of peanut(Arachis hypogaea)allergen by Real-time PCR method with internal amplification control[J].Food Chemistry,2015,174:547-552.
[9]Hoorfar J,Malorny B,Abdulmawjood A,et al.Practical considerations in design of internal amplification controls for diagnostic PCR assays[J].Journal of Clinical Microbiology,2004,42(5):1863-1868.
[10]Malorny B,Hoorfar J,Bunge C,et al.Multicenter validation of the analytical accuracy of Salmonella PCR:towards an international standard.[J].Applied&Environmental Microbiology,2003,69(1):290-296.
[11]Daum LT.Real-time PCR detection of salmonella in suspect foods from a gastroenteritis outbreak in kerr county,Texas.[J]. Journal of Clinical Microbiology,2002,40(8):3050-3052.
[12]Zhang D,Yan Y,Li Q,et al.Label-free and high-sensitive detection of Salmonella using a surface plasmon resonance DNA-based biosensor.[J].Journal of Biotechnology,2012,160(3-4):123-128.
[13]刘斌,史贤明.扩增内标在沙门氏菌PCR检测方法中的应用[J].微生物学通报,2006,33(2):156-161.[14]Singh P,Mustapha A.Development of a real-time PCR melt curve assay for simultaneous detection of virulent and antibiotic resistant Salmonella[J].Food Microbiology,2014,44:6-14.
[15]夏乐先,孙文娟,沈振,等.煮沸裂解法和试剂盒法提取浸矿菌基因组DNA的比较[J].现代生物医学进展,2014(1):31-35,17.
Establishment of a PCR method with an internal amplication control for rapid detection of Salmonlla in foods
ZHANG De-fu1,2,FU Xu-lei1,TANG Yi-wei1,BAI Feng-ling1,YIN Zhe2,YANG Wen-hui2,ZHAO Li-hong1,ZHANG Yi-quan3,LI Chun4,LI Jian-rong1,*
(1.College of Food Science and Project Engineering,Bohai University,Food Safety Key Lab of Liaoning Province,National&Local Joint Engineering Research Center of Storage,Processing and Safety Control Technology for Fresh Agricultural and Aquatic Products,Jinzhou 121013,China;2.Institute of Microbiology and Epidemiology,Academy of Military Medical Sciences,State Key Laboratory of Pathogen and Biosecurity,Beijing 100071,China;3.Schoole of Medicine,Jiangsu University,Zhenjiang 212013,China;4.College of Mathematics and Physics,Bohai University,Jinzhou 121013,China)
A PCR assay was developed for the rapid detection of Salmonlla using 16S rRNA as internal amplification control.Primers were designed to detect the gene of invA in Salmonlla and the assay was optimized and assessment.Detection of various bacterias by PCR indicated that this PCR assay was specific for Salmonlla. The sensitivity of detection for Salmonella purified genomic DNA was 61.1 fg/μL and 2×102cfu/mL for pure cultures.The detection for artificially contaminated egg white showed that Salmonlla could be detected after eight hours enrichment culture when the Sallmonla inoculation was 2 cfu/25 mL.This detection method had a good specificity and sensitivity,and could eliminate false-negative results in the PCR detection method for Salmonella,and suitable for rapid detection of Salmonella in food.
Salmonlla;PCR detection;Internal amplification control;false-negative
TS201.6
A
1002-0306(2015)24-054-05
10.13386/j.issn1002-0306.2015.24.002
2015-04-17
张德福(1983-),男,博士,讲师,研究方向:食品安全,E-mail:zhangdf@bhu.edu.cn。
励建荣(1964-),男,博士,教授,研究方向:水产品和果蔬贮藏加工,食品安全,E-mail:lijr6491@163.com。
“十二五”国家科技支撑计划项目(2012BAD29B06);辽宁省重点实验室开放课题(LNSAKF2013018);辽宁省高等学校创新团队项目(LT2014024);病原微生物生物安全国家重点实验室开放课题(SKLPBS1417,SKLPBS1438);渤海大学博士科研启动项目(BSQD022)。
