In vitro inhibitory effects of plumbagin, the promising antimalarial candidate, on human cytochrome P450 enzymes
2015-10-31WiriyapornSumsakulWannaChaijaroenkulKesaraNaBangchang
Wiriyaporn Sumsakul, Wanna Chaijaroenkul, Kesara Na-Bangchang*
1Graduate Program in Biomedical Sciences, Faculty of Allied Health Sciences, Thammasat University, Pathumthani 12121, Thailand
2Center of Excellence in Pharmacology and Molecular Biology, Graduate Program in Bioclinical Sciences, Chulabhorn International College of Medicine, Thammasat University, Pathumthani 12121, Thailand
In vitro inhibitory effects of plumbagin, the promising antimalarial candidate, on human cytochrome P450 enzymes
Wiriyaporn Sumsakul1, Wanna Chaijaroenkul2, Kesara Na-Bangchang2*
1Graduate Program in Biomedical Sciences, Faculty of Allied Health Sciences, Thammasat University, Pathumthani 12121, Thailand
2Center of Excellence in Pharmacology and Molecular Biology, Graduate Program in Bioclinical Sciences, Chulabhorn International College of Medicine, Thammasat University, Pathumthani 12121, Thailand
ARTICLE INFO
Article history:
in revised form 20 September 2015
Accepted 15 October 2015
Available online 20 November 2015
Metabolism
Human liver microsomes
Plumbagin
Cytochrome P450
Enzyme inhibition
Objective: To investigate the propensity of plumbagin to inhibit the three isoforms of human cytochrome P450 (CYP), ie., CYP1A2, CYP2C19, and CYP3A4 using human liver microsomes in vitro. Methods: Inhibitory effects of plumbagin on the three human CYP isoformswere investigated using pooled human liver microsomes. Phenacetin O-deethylation,omeprazole hydroxylation and nifedipine oxidation were used as selective substrates for CYP1A2, CYP2C19 and CYP3A4 activities, respectively. Concentrations of paracetamol,5-hydroxyomeprazole, and oxidized nifedipine were determined in microsomal incubation mixture using high performance liquid chromatography. Results: Plumbagin showed significantinhibitory effects on all CYP isoforms, but with the most potent activity on CYP2C19-mediated omeprazole hydroxylation. The IC50 (concentration that inhibits enzyme activity by 50%) values of plumbagin and nootkatone (selective inhibitor) for CYP2C19 were(0.78±0.01) and (27.31±0.66) μM, respectively. The inhibitory activities on CYP1A2-mediated phenacetin O-deethylation and CYP3A4-mediated nifedipine oxidation were moderate. The IC50values of plumbagin and -naphthoflavone (selective inhibitor) for CYP1A2 were(1.39±0.01) and (0.02±0.36) μM, respectively. The corresponding IC50values of plumbagin and ketoconazole (selective inhibitor) for CYP3A4 were (2.37±0.10) and (0.18±0.06) μM,respectively. Conclusions: Clinical relevance of the interference of human drug metabolizing enzymes should be aware of for further development scheme of plumbagin as antimalarial drug when used in combination with other antimalarial drugs which are metabolized by these CYP isoforms.
Document heading doi:10.1016/j.apjtm.2015.10.016
1. Introduction
Malaria is widespread in tropical and subtropical regions. The resistance of Plasmodium falciparum (P. falciparum) to first-line antimalarial drugs has resulted in resurgence in treatment failures[1]. Since antimalarial drug resistance compromises the effective treatment of the disease, there is a pressing need for ongoingdrug discovery research that provides effective and affordable antimalarial agents. Natural products including medicinal plants may offer cheap alternative treatment opportunities for malaria patients.
Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone)is the major constituent in several plants including those in Plumbaginaceae, Droseraceae, Ancestrocladaceae, and Dioncophyllaceae families. It is a yellow naphthoquinone pigment which occurs in the plant roots[2]. This compound has been shown to display a wide effect of pharmacological activities such as activities against malaria, leishmaniasis, chagas disease, viral infections, and cancers[2-6]. The ethanol extract of Plumbagoindica Linn. was demonstrated promising antimalarial activity onchloroquine-resistant (K1) and chloroquine-sensitive (3D7)P. falciparum clones with median (range)IC50of 3.0 (2.7-3.1) and 6.2 (6.2-7.3) μg/mL[7]. Furthermore, we have recently demonstrated the antimalarial activity of plumbagin both in vitro and in vivo[8]. The aim of the present study was to further investigate the propensity of plumbagin to inhibit the three isoforms of human cytochrome P450(CYP), ie.,CYP1A2, CYP2C19 and CYP3A4 using human liver microsomes in vitro. The CYP enzyme system plays crucial roles in the metabolism of xenobiotics and endogenous substances and thus,has a significant impact on the occurrence of drug-drug interactions particularly metabolic drug interaction[9]. Interference of hepatic drug metabolizing enzyme(s) of one drug by the co-administered drug may result in unexpectedly high plasma concentration of the affected drug and severe adverse effect or toxicity[10].
2. Materials and methods
2.1. Chemicals
The authentic plumbagin (purity 98.2%) was obtained from Apin chemicals Co. Ltd (OX, UK). Phenacetin, paracetamol, caffeine,omeprazole, 5-hydroxyomeprazole, nifedipine, oxidized nifedipine,ketoconazole, nootkatone, α-naphthoflavone, and diazepam were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1,4-naphthoquinone was purchased from Wako Pure Chemical Industries, Co. Ltd. (Osaka, Japan). β-nicotinamide adenine dinucleotide phosphate (reduced form) tetrasodium salt (NADPH)was purchased from Merck KGaA (Darmstadt, Germany). Pooled human liver microsomes (from 50 donors) were obtained from Gibco BRL Life Technologies (Grand Island, NY, USA).
2.2. CYP inhibition
Inhibitory effects of plumbagin on CYP1A2, CYP2C19 and CYP3A4 activities were investigated in vitro using pooled human liver microsomes in a total volume of 500 μL of 0.1 M sodium phosphate buffer (pH 7.4). The concentration range of each substrate used was approximately equal to its Km (Michaelis constant) value. Each experiment was repeated four times.
2.2.1. HPLC system
Analysis of concentrations of each CYP-mediated metabolite was performed using the validated high-performance liquid chromatography (HPLC) method. The HPLC system consisted of TSP HPLC with P4000 solvent delivery system, equipped with an AS3000 auto sampler, UV1000 detector, SN4000 controller(Thermo Finnigan, San Jose, CA, USA), and Chrome Quest software(version 4.0). The HPLC column used was a Thermo Hypersil Gold C-18 reversed phase column (210 mm×4.6 mm, 5 μm particle size). Quality control (QC) samples were run in duplicate in each analytical batch at low, medium, and high concentrations. Criteria for acceptability were four out of six of the QC analyses to lie inside(100±15)% of the nominal values.
2.2.2. Analytical assay validation
The precision of the assay methods based on intraday repeatability was determined by analyzing five series concentrations of paracetamol, 5-hydroxyomeprazole, or oxidized nifedipine in phosphate buffer. The repeatability between days was established using the same concentration range of the three compounds, but the analysis was performed on three consecutive days. Results are expressed as relative standard deviation (%RSD) of replicate measurements as follow:
The accuracy of the analytical methods was determined by comparing the measured concentration of paracetamol,5-hydroxyomeprazole, and oxidized nifedipine in phosphate buffer at each concentration level (n=5) to the true concentration in three replicates within one day and on three consecutive days. Accuracy was reported as percentage bias calculated from the equation: % Bias = [(Measure value - True value)/True value]×100
Sensitivity of the analytical methods was obtained by the determination the limit of quantification (LOQ). The LOQ was determined based on signal-to-noise approach by comparing measured signals from samples with known lowest concentrations of the test compounds (paracetamol, 5-hydroxyomeprazole, and oxidized nifedipine) and by establishing the minimum concentrations at a typical signal-to-noise ratio is 10:1.
2.2.3. CYP inhibition
The inhibitory effect of plumbagin on CYP1A2-mediated phenacetin O-deethylation was performed using -naphthoflavone as a selective inhibitor[11]. In brief, the reaction mixture was preincubated (at 37 ℃, 5 min) with human liver microsomes (0.3 mg/mL, 100 μL), 20 μM phenacetin, and plumbagin(0-10 μM). The reaction was initiated with the addition of 1 mM NADPH. Following an incubation (at 37 ℃) for 60 min, the reaction was stopped by the addition of 500 μL of cold acetonitrile. The internal standard caffeine (500 μM, 50 μL) was added and the incubation mixture was cooled on ice for 5 min and centrifuged at 12 000× g for 15 min. The supernatant was transferred to an autosampling vial and an aliquot of 20 μL was injected onto the HPLC column. The concentrations of paracetamol (metabolite) were measured by HPLC with UV detection (240 nm)[11]. The gradient mobile phase consisted of a mixture of (A) acetonitrile and (B) distilled water; the initial ratio of mobile phase components (A:B) was 90:10 at a flow rate of 1 mL/min. The calibration curve was plotted using high ratioof paracetamol to caffeine on the ordinate, and concentrations of paracetamol (0.1-50 μM) on the abscissa.
The inhibitory effect of plumbagin on CYP2C19-mediated omeprazole hydroxylation was performed using nootkatone as a selective inhibitor[12]. In brief, the reaction mixture was pre-incubated(at 37 ℃ for 5 min), with human liver microsomes (0.5 mg/mL, 100 μL), 10 μM omeprazole, and plumbagin (0-200 μM). The reaction was initiated with the addition of 1 mM NADPH. Following an incubation (at 37 ℃ for 60 min), the reaction was stopped by the addition of 500 μL of cold acetonitrile. The internal standard 1,4-naphthoquinone (10 μM, 50 μl) was added and the incubation mixture was cooled on ice for 5 min and centrifuged at 12 000×g for 15 min. The supernatant (800 μL) was transferred to an eppendorf tube and evaporated using speed vacuum concentrator (FTS System,Stone Ridge, NY, USA).The dried residue was reconstituted with 100 μL of a mixture of acetonitrile and water (50%:50%, v:v) and 10 μL injected onto the HPLC column. The concentrations of the metabolite 5-hydroxyomeprazole were measured by HPLC with UV detection (302 nm)[12]. The gradient mobile phase consisted of a mixture of (A) acetonitrile and (B) distilled water; the initial ratio of mobile phase components (A:B) was 10:90 at a flow rate of 1 mL/min. The calibration curve was plotted using high ratio of 5-hydroxyomeprazole to 1,4-naphthoquinone on the ordinate,and concentrations of 5-hydroxyomeprazole (40-2 500 nM) on the abscissa.
The inhibitory effect of plumbagin on CYP3A4-mediated nifedipine oxidation was performed using ketoconazole as a selective inhibitor[13]. In brief, the reaction mixture was pre-incubated (at 37 ℃, 5 min), with human microsomes (0.3 mg/mL, 100 μL), 40 μM nifedipine, and plumbagin (0-20 μM). The reaction was initiated with the addition of 1 mM NADPH. Following an incubation (at 37 ℃ for 40 min), the reaction was stopped by the addition of 500 μL of cold acetonitrile. The internal standard diazepam (80 ng/mL,50 μL) was added and the incubation mixture was cooled on ice for 5 min and centrifuged at 12 000×g for 15 min. The supernatant was transferred to an autosampling vial and an aliquot of 20 μL was injected onto the HPLC column. The concentrations of oxidized nifedipine (metabolite) were measured by HPLC with UV detection(270 nm)[13-15]. The gradient mobile phase consisted of a mixture of (A) methanol and (B) distilled water; the initial ratio of mobile phase components (A:B) was 55:45 at a flow rate of 1 ml/min. The calibration curve was plotted using high ratio of oxidized nifedipine to diazepam on the ordinate, and concentrations of oxidized nifedipine (0.1-25 μM) on the abscissa.
2.3. Data analysis
IC50(concentrations causing 50% inhibition of enzyme activity)values were calculated from a logdose—response curve plotted using the Calcusyn™ version 1.1 (BioSoft, Cambridge, UK).Data are presented as mean±SD of the four experiments.
3. Results
3.1. Analytical assay validation
The analytical methods for determination ofparacetamol,5-hydroxyomeprazole, and oxidized nifedipine used in the study was found to be sensitive and accurate. The linearity of all calibration curves were demonstrated with coefficient (r2) of greater than 0.995. CYP1A2-mediated metabolism: The LOQ of paracetamol at a signal-to-noise ratio ≥10 was 100 nM. The precision (intra- and inter-) of analytical method (%RSD) was <6.3% and the accuracy(intra- and inter-) of the method was < ±10% (Table 1).

Table 1 Intra- and inter-assay accuracy and precision of the analytical method for determination of paracetamol (n = 3 for each concentration).
CYP2C19-mediated metabolism: The LOQ of 5-hydroxyomeprazole at a signal-to-noise ratio ≥10 was 40 nM. The precision (intra- and inter-) of analytical method (%RSD) was<6.22% and the accuracy (intra- and inter-)of the method was < ±7%(Table 2).

Table 2 Intra- and inter-assay accuracy and precision of the analytical method for determination of 5-hydroxyomeprazole (n = 3 for each concentration).
CYP3A4-mediated metabolism: The LOQ of oxidized nifedipine at a signal-to-noise ratio ≥10 was 100 nM. The precision (intra- and inter-) of analytical method (%RSD) was <7.05% and the accuracy(intra- and inter-) of the method was < ±6% (Table 3).
3.2. CYPs inhibition
The inhibitory effects of plumbagin and positivecontrols on the activities of three major human CYP isoforms, ie., CYP1A2,CYP2C19 and CYP3A4 are shown in Figure 1A-C and the IC50values are presented in Table 4. Plumbagin clearly inhibited CYP1A2-mediated phenacetin O-deethylation, CYP2C19-mediated omeprazole hydroxylation and CYP3A4-mediated nifedipine oxidation in concentration-dependent manners. Among the three CYP isoforms, the inhibitory activity was most potent for CYP2C19,of which its potency was about 35-fold of the selective inhibitornootkatone (mean IC50values of 0.78 μM vs. 27.31 μM). The inhibitory activities on CYP1A2 and CYP3A4 were moderate (about 13- to 69-fold lower than the selective inhibitors).
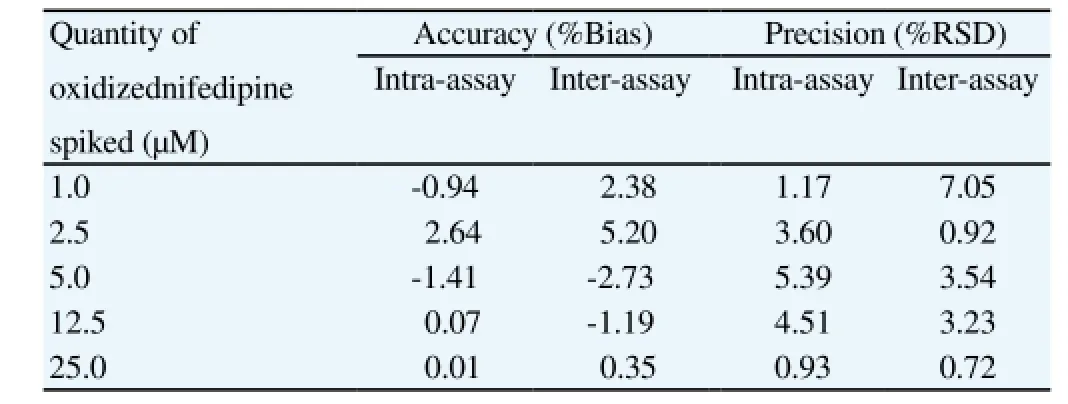
Table 3 Intra- and inter-assay accuracy and precision of the analytical method for determination of oxidized nifedipine (n = 3 for each concentration).
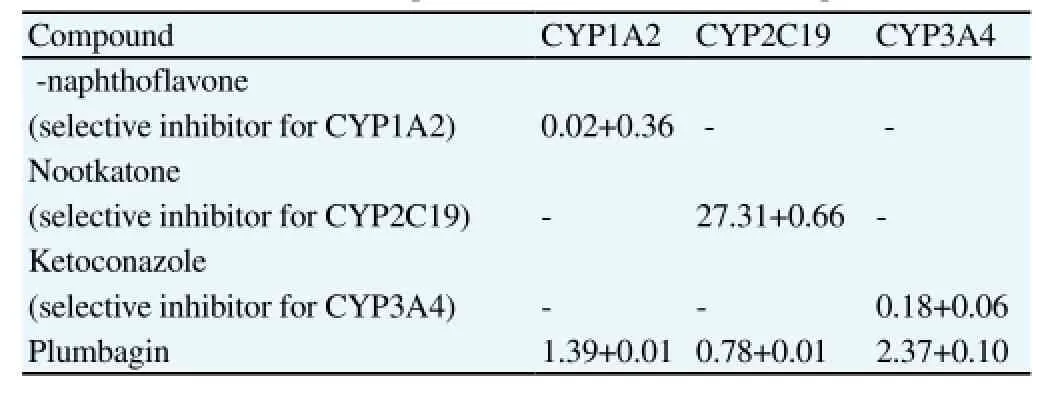
Table 4 IC50values (μM) of plumbagin and selective inhibitors on CYP1A2-mediated phenacetin O-deethylation, CYP2C19-mediated omeprazole hydroxylation,and CYP3A4-mediated nifedipine oxidation (n=4 for each experiment).
4. Discussion
Interactions between phytochemicals in herbal medicines and CYP are now well recognized because of their potential clinical and toxicological implications. These phytochemicals could act as substrates, inhibitors or inducers of the CYP isoforms, which can lead to pharmacokinetic interactions with the co-administered drugs metabolized by the same CYP isoform[9,16]. Our results provide evidence for the inhibitory effect of plumbagin on the three major hepatic CYP isoforms, ie., CYP1A2, CYP2C19, and CYP3A4. CYP1A2, CYP2C and CYP3A are expressed in human liver at approximately 13, 20 and 30% of total CYP, respectively[17]. Specific inhibitors recommended by the US FDA were used as reference compounds for the inhibitory activity on each CYP. For CYP2C19 however, since there has been no recommended selective inhibitor,nootakone was used as a reference compound as its inhibitory activity was shown to be selective toward CYP2C19[12]. Certain extent of variations in inhibitory activities of the selective inhibitors were observed compared with that previously been reported. The discrepancy could be due mainly to the choices of substrates used.
Among the three CYP isoforms under investigation, the inhibitory activity of plumbagin on CYP2C19 was most evident, with potency of about 35-fold of the selective inhibitor nootkatone. CYP2C19 is a major metabolizing enzyme of several clinically important drugs such as proton-pump inhibitors like omeprazole and lanzoprazole,anti-epileptic-like mephenytoin, diazepam, antidepressants, the antiplatelet drug clopidogrel, the antifungal voriconazoleand selective serotonin reuptake inhibitors like citalopram[18]. Previous investigations in man have shown that CYP2C19 activity is susceptible to induction by herbs and natural products, eg., St John's wort, Ginko biloba, and the Chinese herbal mixture Yin Zhi Huang[19-21]. Nevertheless, there has been no clear evidence on the inhibitory effect of herbal remedies on CYP2C19. Although inhibitory activity on CYP3A4 was moderate (mean IC50= 2.37 μM),the clinical relevance of such interaction should not be overlooked as this CYP isoform is involved in the metabolism of 50% of all pharmaceuticals[22]. CYP1A2 is known to play a major role in the metabolism of pre-carcinogens and inhibitory effect of plumbagin to this CYP isoform may contribute only minor interaction with the co-administered drugs[23]. Several other factors are necessary to be considered for definitive conclusion on the clinical relevant metabolic drug interactions. These include comparative dispositionof the individual constituents responsible for inhibition, as well as the locations of the affected CYP (intestine, liver, etc.)[24]. Until further clinical investigations in healthy subjects are confirmed, the potential of this compound for use in treatment of malaria infection may be limited.
The study demonstrated the propensity of plumbagin to interfere with the three human hepatic CYP isoforms, ie., CYP1A2,CYP2C19, and CYP3A4. The inhibitory potency was highest on CYP2C19. Concurrent administration of plumbagin (as pure compound or as the extract of Plumbago indica Linn.) may result in highly toxic plasma concentrations of the co-administered drugs that are metabolized by these CYP isoforms. Clinical relevance of the interference of human drug metabolizing enzymes should be aware of for further development scheme of plumbagin as antimalarial drug when used co-administration with other antimalarial drugs which are metabolized by CYP1A2, 2C19 and 3A4, ie., quinine, mefloquine and chloroquine.
Conflict of interest statement
We declare that we have no conflict of interest.
Acknowledgements
The authors gratefully acknowledge the financial support provided by Thammasat University Research Fund under the TU Research Scholar, Contract No 78/2557, Commission on Higher Education, Ministry of Education of Thailand, Office of Higher Education Commission, Thammasat University (Excellence Center in Pharmacology and Molecular Biology of Malaria and Cholangiocarcinoma), Thammasat University and the Thailand Research Fund through a Royal Golden Jubilee Ph.D. scholarship to Wiriyaporn Sumsakul (Grant no. PHD/0326/2551).
[1] Na-Bangchang K, Congpuong K. Current malaria status and distribution of drug resistance in East and Southeast Asia with special focus to Thailand. Tohoku J Exp Med 2007; 211(2): 99-113.
[2] Paiva SR, Silva Marques S, Figueiredo MR, Auxiliadora M. Plumbaginales: a pharmacological approach. Floresta e Ambiente 2003;10(1): 98-105.
[3] Bhargava SK. Effects of plumbagin on reproductive function of male dog. Indian J Exp Biol 1984; 22(3): 153-156.
[4] Itoigawa M, Takeya K, Furukawa H. Cardiotonic action of plumbagin on guinea-pig papillary muscle. Planta Med 1991; 57(4): 317-319.
[5] Premakumari P, Rathinam K, Santhakumari G. Antifertility activity of plumbagin. Indian J Med Res 1977; 65(6): 829-838.
[6] Sandur SK, Ichikawa H, Sethi G, Ahn KS, Aggarwal BB. Plumbagin(5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-kappaB activation and NF-kappaB-regulated gene products through modulation of p65 and IkappaBalpha kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem 2006; 281(25): 17023-17033.
[7] Thiengsusuk A, Chaijaroenkul W, Na-Bangchang K. Antimalarial activities of medicinal plants and herbal formulations used in Thai traditional medicine. Parasitol Res 2013; 112(4): 1475-1481.
[8] Sumsakul W, Plengsuriyakarn T, Chaijaroenkul W, Viyanant V, Karbwang J, Na-Bangchang K. Antimalarial activity of plumbagin in vitro and in animal models. BMC Complement Altern Med 2014; 14(15).
[9] Zhou S, Gao Y, Jiang W, Huang M, Xu A, Paxton JW. Interactions of herbs with cytochrome P450. Drug Metab Rev 2003; 35(1): 35-98.
[10] Lin JH, Lu AY. Inhibition and induction of cytochrome P450 and the clinical implications. Clin Pharmacokinet 1998; 35(5): 361-390.
[11] Lavhekar S, Lohade A, Coutinho E, Iyer K. Estimation of microsomal CYP1A2 activity by high performance liquid chromatography. Indian J Pharmaceutical Sci 2006; 68(2): 258.
[12] Tassaneeyakul W, Guo L, Fukuda K, Ohta T, Yamazoe Y. Inhibition selectivity of grapefruit juice components on human cytochromes P450. Arch Biochem Biophys 2000; 378(2): 356-363.
[13] Patki K, Von Moltke L, Greenblatt D. In vitro metabolism of midazolam,triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab Dispos 2003; 31(7): 938-944.
[14] Foti RS, Pearson JT, Rock DA, Wahlstrom JL, Wienkers LC. In vitro inhibition of multiple cytochrome P450 isoforms by xanthone derivatives from mangosteen extract. Drug Metab Dispos 2009; 37(9): 1848-1855.
[15] He N, Edeki T. The inhibitory effects of herbal components on CYP2C9 and CYP3A4 catalytic activities in human liver microsomes. Am J Ther 2004; 11(3): 206-212.
[16] Wu JJ, Ai CZ, Liu Y, Zhang YY, Jiang M, Fan XR, et al. Interactions between phytochemicals from traditional Chinese medicines and human cytochrome P450 enzymes. Curr Drug Metab 2012; 13(5): 599-614.
[17] Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals:studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 1994; 270(1): 414-423.
[18] Desta Z, Zhao X, Shin JG, Flockhart DA. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 2002; 41(12): 913-958.
[19] Fan L, Wang G, Wang LS, Chen Y, Zhang W, Huang YF, et al. Herbal medicine Yin Zhi Huang induces CYP3A4-mediated sulfoxidation and CYP2C19-dependent hydroxylation of omeprazole. Acta Pharmacol Sin 2007; 28(10): 1685-1692.
[20] Wang LS, Zhou G, Zhu B, Wu J, Wang JG, El-Aty AMA, et al. St John's wort induces both cytochrome P450 3A4—catalyzed sulfoxidation and 2C19—dependent hydroxylation of omeprazole. Clin Pharmacology & Therapeutics 2004; 75(3): 191-197.
[21] Yin OQ, Tomlinson B, Waye MM, Chow AH, Chow MS. Pharmacogenetics and herb-drug interactions: experience with Ginkgo biloba and omeprazole. Pharmacogenetics 2004; 14(12): 841-850.
[22] Rendic S, Di Carlo FJ. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev 1997; 29(1-2): 413-580.
[23] Ingelman-Sundberg M. Human drug metabolising cytochrome P450 enzymes: properties and polymorphisms. Naunyn Schmiedebergs Arch Pharmacol 2004; 369(1): 89-104.
[24] Izzo AA, Ernst E. Interactions between herbal medicines and prescribed drugs: a systematic review. Drugs 2001; 61(15): 2163-2175.
Article history:
Received 15 August 2015
Received in revised form 20 September 2015
Accepted 15 October 2015
Available online 20 November 2015
15 August 2015
Kesara Na-Bangchang, Center of Excellence in Pharmacology and Molecular Biology of Malaria and Cholangiocarcinoma,Chulabhorn International College of Medicine, Thammasat University (Rangsit Campus), Klongluang, Pathumthani, 12121, Thailand.
E-mail: kesaratmu@yahoo.com
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Demographic, socioeconomic and environmental changes affecting circulation of neglected tropical diseases in Egypt
- Phenolic profile and biological potential of Endopleura uchi extracts
- Roots extracts of Adenophora triphylla var. japonica improve obesity in 3T3-L1 adipocytes and high-fat diet-induced obese mice
- Anti TB drug resistance in Tanga, Tanzania: a cross sectional facility base prevalence among pulmonary TB patients
- Vibrio spp. from Macrobrachium amazonicum prawn farming are inhibited by Moringa oleifera extracts
- Inhibition effect of miR-577 on hepatocellular carcinoma cell growth via targeting β-catenin