Characterisation of a novel, multifunctional, co-processed excipient and its effect on release profile of paracetamol from tablets prepared by direct compression
2015-10-31EragaSylvesterOkhuelegbeArhewohMatthewIkhuoriaUhumwanghoMichaelUwumagbeIwuagwuMagnusAmara
Eraga Sylvester Okhuelegbe, Arhewoh Matthew Ikhuoria, Uhumwangho Michael Uwumagbe, Iwuagwu Magnus Amara
Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, University of Benin, Benin City, 300001, Nigeria
Characterisation of a novel, multifunctional, co-processed excipient and its effect on release profile of paracetamol from tablets prepared by direct compression
Eraga Sylvester Okhuelegbe*, Arhewoh Matthew Ikhuoria, Uhumwangho Michael Uwumagbe, Iwuagwu Magnus Amara
Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, University of Benin, Benin City, 300001, Nigeria
ARTICLE INFO
Article history:
in revised form 1 Jun 2015
Accepted 20 Jun 2015
Available online 5 Aug 2015
Co-processed excipient
Dissolution profiles
Paracetamol tablet
Direct compression
Objective: To characterise a novel multifunctional pharmaceutical excipient and investigate its effect on paracetamol release from tablets prepared by direct compression.
Methods: The excipient was prepared by co-processing gelatinized maize starch with sodium carboxymethyl cellulose and microcrystalline cellulose in a ratio of 2:1:1, dried and pulverized into powder. The excipient formulated was characterized using Fourier transform infrared spectroscopy and differential scanning calorimetry. The excipient was used to prepare batches of tablets by direct compression with drug-excipient ratios of 1:1, 1:2, 1:3 and 1:4. Parameters evaluated on tablets include crushing strength, friability and in vitro dissolution studies.
Results: Differential scanning calorimetry analysis revealed a crystalline excipient while Fourier transform infrared spectroscopy showed no interaction between the excipient and paracetamol. Tablets from all the batches gave average crushing strength values between 3.47 and 4.88 kp. The 1:1 and 1:2 tablet batches were comparable to each other while 1:3 and 1:4 were also comparable to one another in their dissolution profiles. The dissolution parameters of the 1:4 batch was faster with - m∞(90.5%), t50%(3.5 min), t70%(11.6 min) while that of ratio 1:1 was the least with - m∞(48.6%), m5min(23.8%). Their release kinetics followed a Korsmeyer-Peppas model with a super case-II transport mechanism.
Conclusions: The drug-excipient ratios of 1:3 and 1:4 gave pharmaceutically acceptable tablets that met the British Pharmacopoeia specifications. The t50%value of the 1:4 batch of tablets may find its usefulness in formulating drugs for which a fast onset of action is desired.
Original article doi: 10.1016/j.apjtb.2015.07.008 ©2015 by the Asian Pacific Journal of Tropical Biomedicine. All rights reserved.
1. Introduction
The science of drug formulation dates as far back as antiquity,first in the apothecaries and now in modern times, in pharmaceutical industries, compounding pharmacies and research and development laboratories. The aim has always been to formulate a drug product that is acceptable, stable, easy to administer, efficacious, cheap and easy to formulate and that will deliver drug in a desirable manner[1]. In this bid for easier and cheaper methods of formulation,the pharmaceutical scientist is always on the lookout for newer methods and ingredients or excipients that will give a better result. It is important that these newer formulation components do not alter the desired features of the formulation e.g. drug release profile,bioavailability, clinical effectiveness, etc[2].
In the formulation of tablets, the drug and ingredients will needto undergo granulation process to convert them into a state that can be readily compressed into acceptable tablets. However, direct compression (DC) method of tablet manufacture can be used if these formulation components have the necessary degree of fluidity and compressibility[3].
A key component in the DC process is the excipients. They must not only possess those properties which are necessary for satisfactory tablet formulation but retain them when mixed together especially with the active ingredients[4]. Excipients provide a wide variety of functionalities such as better processibility of different active pharmaceutical ingredients into dosage forms, better tablet binding, better tablet disintegration and better active pharmaceutical ingredients bioavailability[5].
The search and development of excipients with better and improved physico-mechanical properties is gaining more attention with the formulation pharmacist and the pharmaceutical companies who are always looking for ways to improve their drug product[6]. No single excipient possesses all the desired physicomechanical properties for the development of a robust drug delivery system. Hence, there is the need to have excipients with multiple characteristics built into them such as better flow, low/no moisture sensitivity, superior compressibility and rapid disintegrationability[7].
The aim of this research work was to formulate and characterize a novel pharmaceutical excipient using the simple method of co-processing and to determine its effect on release profile of paracetamol from tablets.
2. Materials and methods
2.1. Materials
Maize starch British Pharmacopoeia (BP), microcrystalline cellulose (MCC) and sodium carboxymethyl cellulose were gifts from Edo Pharmaceuticals, Edo State, Nigeria, α-lactose monohydrate was obtained from Fluka Chemical Corp., Milwaukee,USA, and paracetamol powder was purchased from Nomagbon Pharmaceuticals, Edo State, Nigeria. All other reagents used were of analytical grade and water was double distilled.
2.2. Excipient preparation
The preparation of the excipient has been previously reported[8]. Six grams of maize starch BP was weighed and dispersed in distilled water at 32 °C, to make a 10 mL slurry in a 500 mL beaker. The slurry was well stirred to ensure that all the powder was properly wetted. Freshly boiled water at 100 °C was then added to the slurry to reach the 200 mL mark and stirred properly till a gel of uniform consistency was formed. Sodium carboxyl methylcellulose (2 g)powder was then dispersed in the gel, in little quantities at a time(to prevent lump formation) and stirred continuously until an even mixture was produced. Then, a mixture containing 2 g of MCC in 5 mL of distilled water was added and stirred until a smooth suspension of all three substances was obtained. The suspension was then transferred into a transparent heat resistant plastic container,spread thinly and dried in the hot air oven at 60 °C for 48 h. The resulting flakes were pulverized using a dry kitchen blender(Phillips, Switzerland) and stored in an air tight container over silica gel until use. The excipient so prepared was termed “Excipient X”.
2.3. Differential scanning calorimetry (DSC) characterization
DSC analysis was carried out using the Netzsch DSC 204 F1 Phoenix apparatus (Netzsch-Geratebau GmbH, Selb, Germany). Four milligrams of the sample was weighed into aluminium pans. The seals were pierced and calibration of the calorimeter was done with indium and the purge gas was nitrogen. Heating of the sample was carried out at the rate of 10 °C per min from 30 to 350 °C under nitrogen at a flow rate of 70 mL/min. The analysis was carried out on paracetamol, maize starch BP, sodium carboxyl methylcellulose,MCC and the tablet granules containing Excipient X.
2.4. Fourier transform infra-red (FTIR) characterization
The FTIR analysis of the samples was carried out using Fourier transform infrared spectrophotometer (Spectrum BX, Perkin Elmer,Beaconsfield, Bucks, England). The potassium bromide pellet method was used; paracetamol powder, components of Excipient X and the tablet granules were scanned at a range of 4 000 - 1 000 cm-1.
2.5. Tablet formulation
Paracetamol tablets were prepared by DC using the formula in Table 1. The different batches of the tablets containing drug-Excipient X ratios of 1:1, 1:2, 1:3 and 1:4 were prepared by weighing the appropriate quantities of paracetamol powder and Excipient X into a mixer and dry mixed for 5 min. Maize starch and lactose were added as disintegrant and filler respectively and the mixer operated for 5 min. The powder mix was compressed into tablets using a single punch tableting machine (F3, Manesty Machine Ltd.,Liverpool, UK) at compression pressure of 32 kilonewton. The die volume was adjusted to compress tablets of uniform weight by using powders weighing 650 mg. The tablets made were then kept in air tight containers and stored in a desiccator until evaluation.
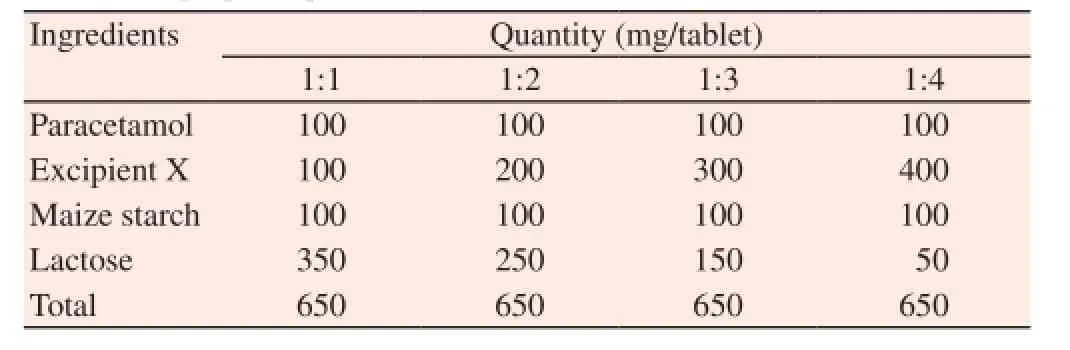
Table 1 Formula of prepared paracetamol tablets.
2.6. Tablet crushing strength
The crushing strength (hardness) of ten tablets per batch was determined by diametral compression using a motorized hardness tester (Campbell Electronics, Model HT-30/50, Mumbai, India). The mean hardness ± standard deviations were calculated.
2.7. Friability test
The weight of ten tablets was determined on an electronic balance(Ohaus Corporation, USA). The tablets were then placed in the drum of a Friabilator (ERWEKA GmbH, Heusenstamm, Germany)revolving at 25 r/min which exposed the tablets to rolling and repeated shock resulting from free fall within the apparatus. After 4 min, the tablets were brought out, de-dusted and reweighed. The weight was then recorded and friability calculated as percentage loss in weight.
2.8. In vitro dissolution test
The dissolution profiles of the paracetamol tablets were determined using the BP basket method for the various batches of the tablets(Caleva ST7, G.B. Caleva Ltd., Dorset, UK). A dissolution medium of 900 mL of 0.1 mol/L HCl solution maintained at (37 ± 0.5) °C with a basket revolution of 50 r/min was used. A 5 mL volume of leaching fluid was withdrawn at predetermined time intervals and replaced with an equivalent volume maintained at the same temperature (37 ± 0.5) °C of the dissolution medium. The samples were filtered and diluted with an equal volume of 0.1 mol/L HCl. This was continued for 60 min. The absorbances of the resulting solutions were measured spectrophotometrically at λmaxof 245 nm(T70, PG Instruments Ltd., Leicestershire, England). The percentage drug released at each time interval was determined using the equation from the standard calibration plot obtained for the pure drug. A minimum of triplicate determinations were carried out for all experiments and the results were reported as mean ± SD.
2.9. Release kinetics
The dissolution data were analysed on the basis of zero order,(cumulative amount of drug released vs time), first order rate (log cumulative amount of drug remaining vs time), Higuchi model(cumulative amount of drug released vs square root of time) and Korsmeyer-Peppas model (log cumulative amount of drug released vs log of time). These are the most frequently reported kinetics of drug release from drug particles and their solid dosage forms[9,10]. The correlation coefficient (r2) for each rate order was calculated. The dissolution profile was considered to follow a particular rate order if the r2value was ≥ 0.99[11].
2.10. Statistical analysis
All data obtained were expressed as mean ± SD. The statistical difference of the paracetamol release data for the batches was accessed by means of the student t-test (P < 0.05) using GraphPad InStat software version 3.36.
3. Results
3.1. Thermal analysis
Figure 1 shows the DSC thermograms of pure paracetamol powder,MCC powder, sodium carboxymethyl cellulose powder, maize starch powder and the tablet granules. Paracetamol thermogram shows a sharp endothermic peak, corresponding to its melting point (169 °C).
3.2. FTIR
The FTIR spectrum of pure paracetamol powder showed characteristic peaks at 1 227.00 cm-1, 1 636.42 cm-1and 3 171.00 cm-1. These peaks observed for paracetamol remained unchanged when compared with the spectral data of the granules (Figure 2).
3.3. Physical parameters of the tablets
Table 2 shows some physical parameters of the paracetamol tablets formulated. All the tablets from all the batches gave mean crushing strength values between 3.47 - 4.88 kp; hardness values greater than or equal to 4 kp is considered to be the minimum for a satisfactory tablet[12]. 3.4. Drug release profile

Table 2 Some physical parameters of the paracetamol tablet.
The release profiles of the various batches of the paracetamol tablets were presented in Figure 3. A comparison of the empirical release data obtained from these curves was presented in Table 3. It can be observed that the batches of tablets of ratio 1:1 and 1:2 gave a comparable release profile with each other while ratios 1:3 and 1:4 also compared with each other (Figure 3).

Table 3 A comparison of the empirical release data of paracetamol tablets.
Correlation coefficients (r2values) are presented in Table 4 which showed that the drug release was most consistent with the Korsemeyer-Peppas model (n > 1), indicating that drug release was essentially by a super case-II transport mechanism[13].

Table 4 Correlation coefficient (r2) and release rate constants (k) of the dissolution studies.
4. Discussion
The sharp peak which appears as a spike is indicative of the purity and crystallinity of paracetamol. On the other hand, the thermogram of the tablet granules containing Excipient X and paracetamol together showed three sharp endothermic peaks with the characteristic peak of pure paracetamol at the middle. Excipient X appears to be more crystalline than amorphous due to the pre-gelled and dried starch component interaction with MCC, which could have facilitated its increase in tablet binding activity and subsequent fast disintegrating action. The broad trough manifested by samples (b),(c) and (d) before 100 °C is probably as a result of loss of moisture. The FTIR spectra ruled out the possibility of chemical interaction and complex formation between paracetamol and Excipient X during the mixing and tableting processes.
The friability values of the tablets increased with decreasing concentrations of Excipient X (Table 2). However, only tablets of two batches (1:3 and 1:4) met the BP specification of a maximum loss of 1% of the mass of the tablets tested or a 0.8% - 1.0% loss in weight of the tested tablets without capping, lamination or breaking up in the course of the test[14,15]. The crushing strength of a tablet can markedly affect the release rate of a drug[16]. Usually, an increase in crushing strength of a tablet is accompanied by a decrease in release rate, due to a decrease in tablet porosity[17]. The change in crushing strength amongst the batches of tablets studied could be the reason for the differences in their release profiles. Though the crushing strength of the 1:4 batch of tablets did not meet the required minimum value of hardness, this can be remedied by increasing the compression pressure of the tablets as long as this does not adversely affect the drug release characteristics of the tablet.
In vitro dissolution data implies that, only batches 1:3 and 1:4 tablets met the BP specification which states that[15], 70% of the uncoated tablet drug should dissolve within 40 min. Another important parameter from the release studies is the t50%value which is very important in formulating drugs that may be needed for fast onset of action. This tells the time at which 50% of the drug was dissolved,which means that in less than 5 min, 50% of the drug from batches 1:3 and 1:4 tablets was already available for absorption.
Excipient X in drug-excipient ratios of 1:3 and 1:4 gave pharmaceutically acceptable tablets that met the BP specifications. The t50%value of the 1:4 batch of tablets may find its usefulness in formulating drugs for which a fast onset of action is desired.
Conflict of interest statement
We declare that we have no conflict of interest.
Acknowledgments
The authors acknowledge the technical support received from the departmental laboratory staff.
[1] Pavliv L, Cahill JF. Formulation and manufacturing. In: Evens RP,editor. Drugs and biological development: from molecule to product and beyond. New York: Springer; 2007, p. 205.
[2] Myrdal PB, Sheth P, Stein SW. Advances in metered dose inhaler technology: formulation development. AAPS PharmSciTech 2014; 15(2): 434-55.
[3] Builders PF, Bonaventure AM, Tiwalade A, Okpako LC, Attama AA. Novel multifunctional pharmaceutical excipients derived from microcrystalline cellulose-starch microparticulate composites prepared by compatibilized reactive polymer blending. Int J Pharm 2010;388(1-2): 159-67.
[4] Armstrong NA. Tablet manufacture. In: Swarbrick J, Boylan JC, editors. Encyclopedia of pharmaceutical technology. 2nd ed. New York: Marcel Dekker Inc.; 2002, p. 2713-32.
[5] Adeoye O, Alebiowu G. Flow, packing and compaction properties of novel coprocessed multifunctional directly compressible excipients prepared from tapioca starch and mannitol. Pharm Dev Technol 2014;19(8): 901-10.
[6] Block LH, Moreton RC, Apte SP, Wendt RH, Munson EJ, Creekmore JR. Co-processed excipients. Pharmacopeial Forum 2009; 35(4): 1026-28.
[7] ElShaer A, Hanson P, Mohammed AR. A systematic and mechanistic evaluation of aspartic acid as filler for directly compressed tablets containing trimethoprim and trimethoprim aspartate. Eur J Pharm Biopharm 2013; 83(3): 468-76.
[8] Eraga SO, Damisah CO, Uhumwangho MU, Iwuagwu MA. Development and evaluation of novel, multifunctional co-processed excipients for direct compression of paracetamol tablets. J Sci Pract Pharm 2014; 1(1): 25-30.
[9] Perge L, Robitzer M, Guillemot C, Devoisselle JM, Quignard F, Legrand P. New solid lipid microparticles for controlled ibuprofen release: formulation and characterization study. Int J Pharm 2012; 422(1-2): 59-67.
[10] Nkansah P, Antipas A, Lu Y, Varma M, Rotter C, Rago B, et al. Development and evaluation of novel solid nanodispersion system for oral delivery of poorly water-soluble drugs. J Control Release 2013;169(1-2): 150-61.
[11] Iqbal Z, Khan R, Nasir F, Khan JA, Rashid A, Khan A, et al. Preparation and in-vitro in-vivo evaluation of sustained release matrix diclofenac sodium tablets using PVP-K90 and natural gums. Pak J Pharm Sci 2011;24(4): 435-43.
[12] Rudnic EM, Schwartz JD. Oral solid dosage forms. In: Gennaro AR,editor. Remington: the science and practice of pharmacy. 20th ed. Philadelphia: Lippincot Williams and Wilkins Inc.; 2000, p. 858-93.
[13] Abioye AO, Kola-Mustapha A. Formulation studies on ibuprofen sodium-cationic dextran conjugate: effect on tableting and dissolution characteristics of ibuprofen. Drug Dev Ind Pharm 2015: 1-21. doi:10.31 09/03639045.2015.1024684.
[14] Zimmer L, Kasperek R, Poleszak E. [Application of β-cyclodextrin in the formulation of ODT tablets containing ibuprofen]. Polim Med 2014;44(4): 231-5. Polish.
[15] The British Pharmacopoeia. London: The Pharmaceutical Press; 2009.
[16] Adeleye OA, Femi-Oyewo MN, Odeniyi MA. The effect of processing variables on the mechanical and release properties of tramadol matrix tablets incorporating Cissus populnea gum as controlled release excipient. Polim Med 2014; 44(4): 209-20.
[17] Keny RV, Desouza C, Lourenco CF. Formulation and evaluation of rizatriptan benzoate mouth disintegrating tablets. Indian J Pharm Sci 2010; 72(1): 79-85.
17 Apr 2015
Eraga Sylvester Okhuelegbe, Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, University of Benin, Benin City, 300001, Nigeria.
Tel: +2348030884928
E-mail: eragaso@uniben.edu
杂志排行

Asian Pacific Journal of Tropical Biomedicine的其它文章
- Leishmaniosis phytotherapy: Review of plants used in Iranian traditional medicine on leishmaniasis
- Isolation, screening and identification of Bacillus spp. as direct-fed microbial candidates for aflatoxin B1biodegradation
- Morphological and molecular characterization of fungus isolated from tropical bed bugs in Northern Peninsular Malaysia, Cimex hemipterus (Hemiptera: Cimicidae)
- Effects of filaricidal drugs on longevity and enzyme activities of the microfilariae of Setaria cervi in white rats
- Dill tablet: a potential antioxidant and anti-diabetic medicine
- Plantago major treatment enhanced innate antioxidant activity in experimental acetaminophen toxicity