反相高效液相色谱法测定心脉康合剂中淫羊藿苷含量
2015-10-26白荣龚小倩邱海蕴陶恩
白荣,龚小倩,邱海蕴,陶恩
(1.三峡大学仁和医院,湖北宜昌443001;2.三峡食品药品检验检测中心,湖北宜昌443000;3.湖北民族学院附属民大医院,湖北恩施445000)
反相高效液相色谱法测定心脉康合剂中淫羊藿苷含量
白荣1,龚小倩2,邱海蕴2,陶恩3
(1.三峡大学仁和医院,湖北宜昌443001;2.三峡食品药品检验检测中心,湖北宜昌443000;3.湖北民族学院附属民大医院,湖北恩施445000)
目的建立测定心脉康合剂中淫羊藿苷含量的反相高效液相色谱法,为质量控制提供依据。方法色谱柱为Diamonsil C18柱(250mm×4.6mm,5μm),流动相为乙腈-0.05%磷酸溶液(25∶75),流速为1.0mL/min,检测波长为270 nm。结果淫羊藿苷进样量线性范围为0.062 6~1.879 8μg(r=1.000 0),精密度、稳定性及重复性的RSD均小于2.00%,平均回收率为99.35%,RSD为0.79%(n=9)。结论该方法简便、准确、重复性好,可用于该制剂质量控制。
心脉康合剂;淫羊藿苷;反相高效液相色谱法
心脉康合剂是由麦冬、丹参、淫羊藿、党参、附片、五味子、黄芪、甘草、桂枝、菟丝子、赤芍11味中药组方的中药复方制剂,临床用于脾肾两虚引起的气血虚弱、心阳不足及冠心病、心肾功能不全、免疫功能失调。该制剂在我院临床已应用多年,效果良好。淫羊藿主要功能为补肾阳、强筋骨,用于肾阳虚衰、阳痿遗精。在目前的心脉康合剂质量标准中,只有对麦冬、丹参的薄层色谱鉴别,未对其中任一药材的有效成分进行定量分析。为提高医院制剂质量标准,有效控制剂质量,参考文献[1-7],建立了心脉康合剂中淫羊藿苷含量测定的反相高效液相色谱(RP-HPLC)法,现报道如下。
1 仪器与试药
Waters e2695型高效液相色谱仪,Empower色谱工作站(美国Waters公司);METTLER-AE240型电子分析天平。淫羊藿苷对照品(中国药品生物制品检定所,供含量测定用,批号为110737-200415);乙腈为色谱纯,水为去离子双蒸馏水,其他试剂均为分析纯;心脉康合剂(湖北民族学院附属医院制剂室,批号为140109,140308,140809);缺淫羊藿的阴性对照合剂(自制)。
2 方法与结果
2.1色谱条件及系统适用性试验
色谱柱:Diamonsil C18柱(250mm×4.6mm,5μm);流动相:乙腈-0.05%磷酸溶液(25∶75);流速:1.0mL/min;检测波长:270 nm;柱温:25℃;进样量:20μL。在此色谱条件下,淫羊藿苷达到基线分离(分离度大于1.5),理论板数按淫羊藿苷峰计算应不低于3 000。色谱图见图1。
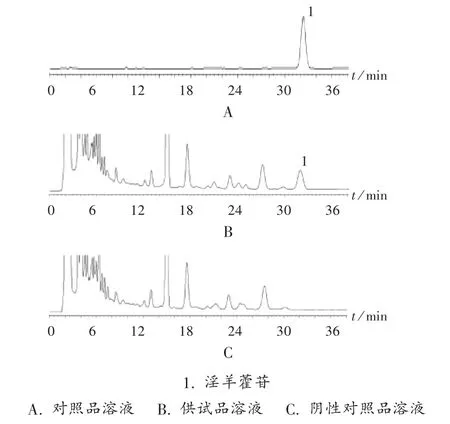
图1 高效液相色谱图
2.2溶液制备
精密称取60℃减压干燥14 h后的淫羊藿苷对照品19.57mg,置25mL容量瓶中,用甲醇溶解并稀释至刻度,作为贮备液;精密量取贮备液2 mL,置25mL容量瓶中,加甲醇溶解并稀释至刻度,摇匀,即得质量浓度为62.62μg/mL的对照品溶液。精密量取供试品5mL,置50 mL容量瓶中,加甲醇约45 mL,超声溶解15min,放置至室温,加甲醇至刻度,摇匀,滤过,取续滤液,即得供试品溶液。取处方量药材(不含淫羊藿)按心脉康合剂生产方法制备缺淫羊藿的阴性对照合剂3批,混合均匀,作为阴性对照品溶液。
2.3方法学考察
专属性试验:分别精密量取对照品溶液、供试品溶液、阴性对照品溶液各20μL,在拟订色谱条件下分别注入色谱仪,记录色谱图,见图1。供试品溶液色谱中,在与对照品溶液色谱相同位置上有相同时间的保留峰,阴性对照品溶液色谱则无此峰。淫羊藿苷保留时间约为32.4min,理论板数以淫羊藿苷峰计算为12500。
线性关系考察:分别精密量取按2.2项下对照品溶液1.0,5.0,10.0,20.0,30.0μL,注入色谱仪,以色谱峰面积(Y)为纵坐标、进样量(X,μg)为横坐标进行线性回归,得回归方程Y=2 306 468X-37 400,r=1.000 0(n=5)。结果,淫羊藿苷进样量在0.062 6~1.879 8μg范围内与峰面积线性关系良好。
精密度试验:取同一对照品溶液,重复进样6次,每次10μL,记录峰面积。结果峰面积平均值为1 406 601,RSD为0.2%(n=6),表明仪器精密度良好。
重复性试验:取同一批样品(批号140109)6份,按2.2项下方法制备供试品溶液,进样20μL,依法测定。结果合剂中淫羊藿苷含量分别为0.432,0.429,0.427,0.430,0.429,0.435 g/L,平均0.430 g/L,RSD为0.7%(n=6),表明方法重复性好。
稳定性试验:取同一供试品溶液,分别于0,1,4,12,24,48,72 h时进样20μL,依法测定。结果淫羊藿苷峰面积的RSD为1.0%(n=7),表明供试品溶液至少在3 d内稳定。
检测限与定量限确定:精取对照品溶液,用甲醇逐级稀释,记录色图谱。信噪比为3时测得检测限为2.50 ng,信噪比为10时测得定量限为12.52 ng。
加样回收试验:精密量取2mL已知含量的样品9份(批号140109),分别精密加入按2.2项下方法制备的对照品贮备液0.5,2.0,4.0mL各3份,再按样品测定项下方法配制成供试品溶液50mL,进样20μL测定,计算回收率。结果见表1。
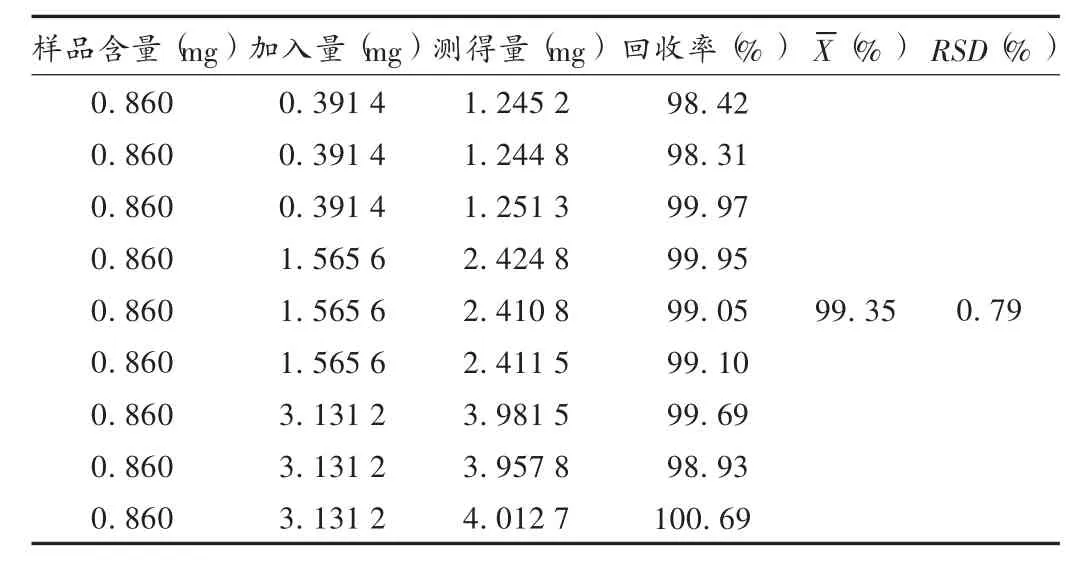
表1 淫羊藿苷加样回收试验结果(n=9)
2.4样品含量测定
取3批样品,按2.2项下方法制备供试品溶液。分别精密量取对照品溶液和供试品溶液各20μL,按色谱条件进样测定,记录峰面积,计算样品中淫羊藿苷的含量。结果批号分别为140109,140308,140809的样品含量分别为0.430,0.427,0.434 g/L,平均含量为0.430 g/L。
3 讨论
淫羊藿作为主要组分广泛应用于多种制剂中,在本合剂组方中,淫羊藿即为主要成分。淫羊藿苷是淫羊藿中的主要成分,也是其主要活性成分之一,测定其含量对于控制本品质量,更好地保证临床疗效有重要意义。
淫羊藿检测方法比较成熟,对其定量检测通常采用高效液相色谱(HPLC)法,含淫羊藿中成药或制剂中淫羊藿苷的HPLC法含量测定报道已较多[1-7],主要流动相为甲醇-水(磷酸溶液)、乙腈-水(磷酸溶液),但因各制剂处方组成差异,流动相比例各不相同,采用的洗脱方式也多种多样,以充分消除样品中其他成分的干扰。本研究中在建立心脉康合剂中淫羊藿苷含量测定时,对药典及文献中报道的方法进行了研究测试,最终选定流动相组成、磷酸溶液浓度、比例及洗脱方式,筛选出的色谱条件对本制剂中淫羊藿苷的测定具有较强的专属性,能达到基线分离、峰形对称、阴性对照无干扰,不足之处可能在于洗脱时间较长,但简便的样品处理方式抵消了这种不足。
为了消除中药制剂中主要成分测定过程中的干扰,提高目标检测物的可检测性,中药制剂样品处理方式非常重要。含淫羊藿固体制剂的前处理方法大多为超声提取[4-7],其次是回流提取[4],对液体制剂多直接用溶剂稀释[4]。由于本制剂为液体制剂,不存在被检成分能否完全溶出的问题,因此在样品制备时首先选择了操作简便的直接溶剂稀释法。研究表明,取适量合剂样品进行适当稀释后,在线性范围内可对合剂中的淫羊藿苷进行准确检测,无需浓缩或提取过程[4]。
综上所述,本研究中建立的淫羊藿苷含量测定方法简便、快速、结果准确、重复性好,可用于心脉康合剂的质量控制。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2010:306.
[2]邢婧,任红,汪轩,等.HPLC法同时测定淫羊藿-川芎药对8种化学成分的含量[J].药物分析杂志,2015,35(6):960-965.
[3]杨炳川,杨浩然,方应权,等.HPLC法测定复方玄驹胶囊中淫羊藿苷的含量[J].中国药房,2015,35(6):1 282-1 283.
[4]黄朝情,彭玉德,黄文华,等.12种中成药中淫羊藿组分的质量考察[J].中国药房,2011,22(43):4 086-4 088.
[5]刘源,石继伟.高效液相色谱法测定安神补脑片中淫羊藿苷含量[J].中国药业,2014,23(1):14-15.
[6]鲍蕾蕾,陈海飞,卞俊,等.高效液相色谱法测定健步关节胶囊中淫羊藿苷含量[J].中国药业,2012,21(20):42-43.
[7]王巨存,冯鑫,赵薇,等.HPLC法测定壮骨止痛颗粒中淫羊藿苷的含量[J].药物分析杂志,2011,31(4):742-744.
Determ ination of Icariin in Xinmaikang M ixture by RP-HPLC
Bai Rong1,Gong Xiaoqian2,Qiu Haiyun2,Tao En3
(1.Renhe Hospital of Three Gorges University,Yichang,Hubei,China 443001;2.Three Gorges Center for Food and Drug Control,Yichang,Hubei,
China 443000;3.The Affiliated Hospital of Hubei University for Nationalties,Enshi,Hubei,China 445000)
Objective To establish a reverse phase-high performance liquid chromatography(RP-HPLC)method for the determination of icariin in Xinmaikang Mixture,so as to provide
for its quality control.M ethods The HPLC analysis was carried out on the Diamonsil C18column(250 mm×4.6 mm,5μm).The mobile phase was consisted of acetonitrile-0.05%phosphoric acid(25∶75)with the flow rate of 1.0 mL/min.The detection wavelength was 270 nm.Results The linear range of icariin was 0.062 6μg~1.879 8μg(r=1.000 0),the RSDs of precision,stability and reproducibility tests were lower than 2.00%,with an average recovery rate of icariin 99.35%,RSD=0.79%(n=9).Conclusion This method is sensitive,simple and accurate with good repeatability and good linearity.It can be used for the quality contnet of icariin in Xinmaikang Mixture.
Xinmaikang Mixture;icariin;reverse phase-high performance liquid chromatography
R284.1;R286.0
A
1006-4931(2015)24-0158-03
白荣(1962-),女,大学本科,副主任药师,研究方向为医院药学、药物分析及药物制剂,(电子信箱)brlk933@ 163.com。
2015-08-13)
