绿色木霉TvALP-linker-TvRBL融合基因的构建与原核表达
2015-10-25徐杨玉温世杰李海芬李玲梁炫强
徐杨玉温世杰李海芬李玲梁炫强
(1.广东省农业科学院作物研究所,广州 510640;2.华南师范大学生命科学学院,广州 510631)
绿色木霉TvALP-linker-TvRBL融合基因的构建与原核表达
徐杨玉1,2温世杰1李海芬1李玲2梁炫强1
(1.广东省农业科学院作物研究所,广州 510640;2.华南师范大学生命科学学院,广州 510631)
在TvALP基因和TvRBL基因间加入一段柔性链接头(linker)基因序列,构建融合基因。生物信息学分析表明该融合基因含有一段信号肽序列。利用RT-PCR技术从绿色木霉菌丝体总RNA中扩增出TvRBL基因、TvALP基因和去除信号肽的ΔTvALP基因,分别克隆到原核表达载体pET30a,构建重组质粒pET30a-TvALP-linker-TvRBL和pET30a-ΔTvALP-linker-TvRBL,转化大肠杆菌BL21(DE3)pLysS,经异丙基硫代-β-D-半乳糖苷(IPTG)诱导表达。SDS-PAGE电泳分析结果表明,去除信号肽的表达质粒pET30a-ΔTvALP-linker-TvRBL在大肠杆菌BL21(DE3)pLysS中获得了表达,在60 kD处有一条蛋白质特异条带,与预测的目的产物蛋白条带大小一致。
绿色木霉;TvALP-linker-TvRBL融合基因;信号肽;原核表达
生物防治黄曲霉毒素污染是目前国内外研究的热点、难点。绿色木霉是重要的生物防治菌,在植病生防中具有重要的作用。它的作用机制有以下几种:(1)抗生作用;(2)重寄生作用;(3)溶菌作用;(4)竞争作用(主要为对生存空间和营养的竞争);(5)诱导植物抗性;(6)协同拮抗作用;(7)毒性蛋白。
绿色木霉中的蓖麻毒素-B-凝集素(TvRBL)和碱性蛋白酶(TvALP)在植病生防中的作用机制分别属于毒性蛋白和重寄生作用,其中重寄生作用是主要的防治机制。碱性蛋白酶可以降解植物病原真菌细胞壁,从而抑制病原菌的孢子萌发,并引起菌丝和孢子的崩解,以达到杀灭病原真菌的目的[1-4]。1978 年Rodriguez-Kabana发现蛋白酶活性与绿色木霉(T. viride)防治白绢病(S. rotlfsii)的能力有关[5]。而蓖麻毒蛋白-B-凝集素最早在伞菌中发现具有血凝活性[6],其细胞凝集作用机制是凝集素分子的一个亚基与一个细胞表面的凝集素专一识别的糖基结合,另一个亚基与另一个细胞上的糖基结合,从而通过凝集素的“架桥”作用而导致的[7]。1994年Pemberton[8]发现他所研究的400余种大型真菌中,50%具有很强的凝集素活性。
本研究前期成功克隆了绿色木霉TvRBL和TvALP两个基因,并在大肠杆菌中诱导表达,TvRBL蛋白为可溶性表达;而TvALP蛋白形成了包涵体,复性不成功,无法研究对黄曲霉的作用机理,因此获得有活性的碱性蛋白酶对生物防治黄曲霉意义重大。目前,构建融合蛋白已经成为一种行之有效的方法来增加可溶性蛋白的表达和方便蛋白的纯化[9,10]。其方法之一是通过连接肽序列将功能分子连接起来。本研究通过添加5个中性氨基酸的柔性多肽序列将TvRBL基因连接到TvALP基因的下游,构建pET30a-TvALP-linker-TvRBL以及切除信号肽的pET30a-ΔTvALP-linker-TvRBL原核表达载体,切除信号肽后获得目的蛋白,旨在为进一步研究该融合蛋白的可溶性表达以及抗黄曲霉污染的分子机制奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株与载体质粒 菌株绿色木霉(Trichoderma viride)由本实验室前期获得;黄曲霉(Aspergillus flavus L.)菌株GIM3.493引自中国科学院微生物研究所,该菌株侵染能力和产毒能力均较强;大肠杆菌DH5α、BL21(DE3)pLysS是本实验室保存的菌种;载体有pUM-T Vector(Bioteke)、pET30a Vector(Novagen)。
1.1.2 试剂与工具酶盒 丝状真菌RNA提取试剂盒、纯化试剂盒购自Omega公司;各限制性内切酶、T4连接酶、DNA Marker、反转录试剂盒M-MLV RTase cDNA Synthesis Kit购自TaKaRa公司;PCR引物合成与测序由Invitrogen公司完成;BioTeke 2×Power Taq PCR Master Mix购Bioteke;蛋白Marker、Gel Extraction Kit均为TIANGEN公司的产品;质粒提取试剂盒购自Axygen公司;氨苄青霉素、卡那霉素、氯霉素等购自北京鼎国生物技术发展中心;SDS、Tris、丙烯酰胺、甲叉双丙烯酰胺、β-巯基乙醇、考玛斯亮蓝R-250、TEMED、过硫酸胺、二硫苏糖醇(DTT)等常用试剂均为国产分析纯。
1.1.3 常用溶液及培养基 LB 液体培养基(g/L):酵母膏5、胰蛋白胨10、NaCl 10,加过柱水,调pH值至 7.0,常压灭菌20 min;LB 固体LB培养基在液体LB培养基基础上加15 g琼脂粉。
绿色木霉菌丝基础培养基:Mandels营养盐浓缩液100 mL,Mandels微量元素浓缩液1 mL,1 mol/L的柠檬酸缓冲液(pH4.5)50 mL,吐温-80 2 mL,胰蛋白胨1.5 g,D-葡萄糖20 g,双蒸水定容至1 L。PDA培养基:马铃薯200 g,蔗糖20 g,琼脂20 g,蒸馏水1 L。把马铃薯去皮后,切成1 cm3的小块煮沸30 min,然后用纱布过滤,再加蔗糖和琼脂,并补足水至1 L,混匀后分装,115℃灭菌20 min,室温保存。
50×TAE:242 g Tris,37.2 g Na2EDTA·2H2O,800 mL去离子水溶解,加入57.1 mL醋酸,混匀,去离子水定容至1 L,室温保存备用。
常用抗生素:氯霉素用无水乙醇溶解,其余抗生素灭菌水溶解,0.22 μm滤膜过滤,分装,-20℃保存备用。
1.2 方法
1.2.1 融合基因的生物信息学分析 根据实验室前期克隆获得的TvALP cDNA基因序列(GenBank登录号:KJ659907)和TvRBL cDNA基因序列(GenBank登录号:KJ659907),去除TvALP基因的终止密码子和TvRBL基因的起始密码子,中间用5个中性氨基酸的小型连接(GGCGGTGGCGGCAGC),将TvRBL基因连接到TvALP基因的下游,构建TvALP-linker-TvRBL融合基因(图1)。生物信息学分析(图2)表明,该融合基因前20个氨基酸为信号肽序列。
1.2.2 PCR引物与linker的设计 根据预测的融合基因序列,运用Primer 5软件设计并合成特异性引物。扩增TvALP基因序列的引物为Tvalp;扩增ΔTvALP基因序列的引物为ΔTvalp;扩增TvRBL基因序列的引物为Tvrbl。具体的PCR引物序列见表1。

图1 TvALP-linker-TvRBL融合基因

图2 TvALP-linker-TvRBL融合基因信号肽的预测

表1 PCR引物设计
1.2.3 目的片段的扩增、PCR鉴定与测序 以绿色木霉菌丝体总RNA反转录得到的cDNA为模板,使用引物Tvalp、ΔTvalp及Tvrbl,分别扩增TvALP、ΔTvALP和TvRBL三个目的片段,PCR反应体系:2×PCR Master Mix 10 μL;引物各1.0 μL;模板为1.0 μL,加双蒸水至总体积20 μL。PCR程序为:95℃预变性5 min;95℃变性30 s,55℃退火30 s,72℃延伸1 min,35个循环;最后72℃延伸10 min。反应结束后,1%琼脂糖凝胶电泳检测。从琼脂糖凝胶上切下含目的片段的凝胶,按照DNA凝胶回收试剂盒操作纯化回收扩增产物。
回收纯化后的目的片段与pUM-T质粒连接调制成10 μL,反应体系如下:pUM-T质粒1 μL,T4 DNA Ligase(3 U/μL)1 μL,2×T4 DNA Rapid Ligation Buffer 5 μL,目的片段3 μL,22-25℃反应10-30 min。取连接产物10 μL加入冰上融化的100 μL感受态细胞DH5α,混匀冰浴30 min;42℃热激90 s,冰浴2 min。向离心管中加入500 μL 37℃预热的(不含抗生素)LB培养基150 r/min、37℃振荡培养60 min,使质粒上相关的抗性标记基因表达;取100 μL菌液涂布于含抗生素的筛选平板上,正面向上放置30 min,待菌液完全被培养基吸收后倒置培养皿,37 ℃培养过夜。
挑取LB平板培养基上的孤立菌落,以1 μL菌悬液为模板,PCR反应体系和程序同上,将预变性时间延长到15 min。反应结束后1%的琼脂糖电泳检测,挑选阳性克隆测序。测序正确的重组克隆质粒命名为pUM-T-TvALP、pUM-T-ΔTvALP及pUM-TTvRBL。
1.2.4 融合基因重组表达质粒的构建与鉴定 Kpn I和BamHⅠ双酶切碱裂解法获得的pUM-T-TvALP质粒、pUM-T-ΔTvALP质粒和pET30a载体,连接转化大肠杆菌DH5α,挑取单克隆,37℃过夜摇菌,碱裂解法提质粒,进行PCR和双酶切鉴定,测序鉴定正确的重组质粒命名为pE30a-TvALP和pE30a-ΔTvALP。
BamHⅠ和Hind Ⅲ双酶切碱裂解法获取的pUM-T-TvRBL重组质粒和鉴定正确的pET30a-TvALP、pET30a-ΔTvALP重组质粒,连接转化BL21(DE3)pLysS菌株感受态细胞,涂布于含有终浓度50 μg/mL Kanamycn和34 μg/mL Chloromycetin的LB选择性平板上,37℃培养,挑取单菌落,进行PCR鉴定。以构建好的重组质粒pET30a-TvALP-linker-TvRBL和pET30a-ΔTvALP-linker-TvRBL为 模板,用Tvalp、Tvalp-Tvrbl、Tvrbl、ΔTvalp、ΔTvalp-Tvrbl这5对引物分别进行PCR扩增,扩增产物用1%的琼脂糖凝胶电泳检测。用Kpn Ⅰ 和BamHⅠ、BamHⅠ和Hind Ⅲ、Kpn Ⅰ和Hind Ⅲ分别对两个重组质粒进行双酶切,酶切产物经1%的琼脂糖电泳检测。测序鉴定正确的重组质粒命名pET30a-TvALP-linker-TvRBL、pET30a-ΔTvALP-linker-TvRBL。
1.2.5 融合蛋白的诱导表达 提取质粒pET30a-TvALP-linker-TvRBL、pET30a-ΔTvALP-linker-TvRBL分别转化大肠杆菌BL21(DE3)pLysS,分别挑取一个单克隆菌落接种于含有终浓度为50 μg/mL Kanamycn和34 μg/mL Chloromycetin 的5 mL LB液体培养基中,37℃培养过夜至对数生长期。次日按1%转接扩大培养,当OD600达到0.6左右时,加入IPTG至终浓度为1 mmol/L,37℃进行化学诱导表达。每隔2 h取样,继续诱导6 h,同时以转化有空载体pET30a的BL21(DE3)pLysS为阴性对照。分别离心收集细菌沉淀,每管加入400 μL蛋白上样缓冲液,重悬后煮沸10 min,10 000×g离心10 min取上清液备用。
SDS-PAGE分析:配制12%分离胶和5%浓缩胶,取上述样品,每孔30 μL,浓缩胶电压90 V,分离胶电压120 V,电泳结束后,参照康彬等[11](2000)的方法,剥胶后用ddH2O冲洗数次,在考马斯亮蓝染色液中短暂振动染色20 min,然后用ddH2O反复冲洗后,浸入250 mmol/L KCl溶液中,直到背景清晰为止,整个过程大约需要1 h。暂时保存在保存液中或立即拍照观察。
2 结果
2.1 目的片段的扩增
琼脂糖凝胶电泳鉴定结果(图3)表明,以绿色木霉菌丝体总RNA反转录得到的cDNA为模板,使用P1和P2、P1'和P2、P3和P4三对基因特异性引物,进行RT-PCR扩增,结果获得 TvALP、ΔTvALP和TvRBL三个目的片段,大小约1 227、1 167及456 bp,与预期大小相符。

图3 PCR产物的琼脂糖凝胶电泳分析
2.2 重组克隆质粒的构建与鉴定
通过对pUM-T-TvALP、pUM-T-ΔTvALP和pUMT-TvRBL重组克隆质粒的菌液PCR鉴定,分别得到大小约1 227、1 167及456 bp的目的片段(图4),结果均与预期值相符,视为拟阳性克隆,送华大测序,验证正确后,表明目的基因均已插入载体质粒pUM-T多克隆位点。

图4 重组克隆质粒的菌落PCR检测
2.3 融合基因重组表达质粒的构建与鉴定
通过对重组质粒pET30a-TvALP和pET30a-ΔTv-ALP的Kpn Ⅰ 和BamH Ⅰ双酶切鉴定、PCR鉴定,分别得到大小约5 382、1 227和1 167 bp的目的片段(图5,图6),结果均与预期值相符,证明质粒中含有目的片段,并且基因序列和阅读框架均正确,表明已成功构建了pET30a-TvALP和pET30a-ΔTvALP载体。
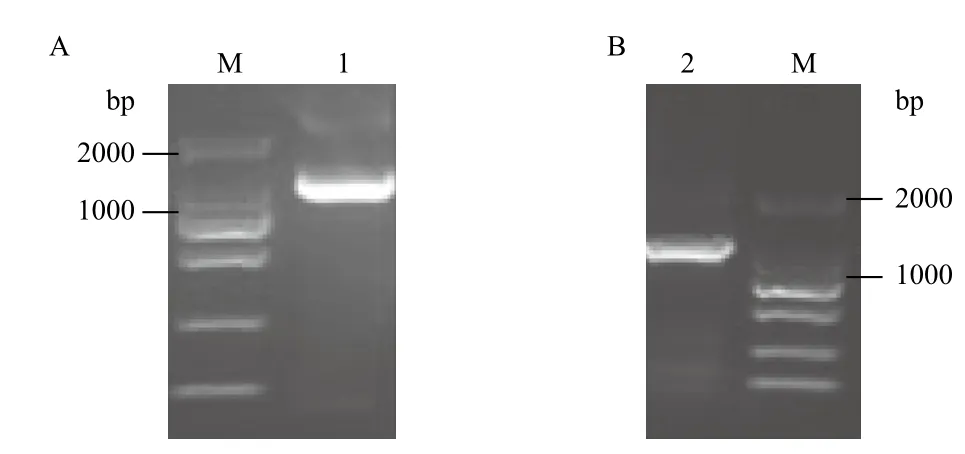
图5 重组表达质粒pET30a-TvALP(A)和pET30a-ΔTvALP(B)的PCR鉴定

图6 重组表达质粒pET30a-TvALP(A)和pET30a-ΔTvALP(B)的双酶切鉴定
通过对重组质粒pET30a-TvALP-linker-TvRBL和pET30a-ΔTvALP-linker-TvRBL的BamH Ⅰ和HindⅢ双酶切和PCR鉴定,分别得到大小约为5 377、1 227、456、1 683、5 377、1 167、456和 1 623 bp的目的片段(图7,图8),结果均与预期值相符,证明两个质粒中分别含有TvALP-linker-TvRBL以及ΔTvALP-linker-TvRBL融合基因,并且基因序列和阅读框架均正确,表明已成功构建了TvALP-linker-TvRBL以及ΔTvALP-linker-TvRBL融合基因的表达载体。

图7 重组表达质粒pET30a-TvALP-linker-TvRBL的PCR(A)和双酶切(B)鉴定

图8 重组表达质粒pET30a-ΔTvALP-linker-TvRBL的PCR(A)和双酶切(B)鉴定
2.4 融合蛋白的诱导表达
含重组质粒pET30a-TvALP-linker-TvRBL的菌株未能诱导出蛋白,但含重组质粒pET30a-ΔTvALP-linker-TvRBL的菌株在60 kD处出现一条新生蛋白带(图9,图10),与预测的融合蛋白大小相近,表明切除信号肽后可以诱导融合蛋白的表达。

图9 TvALP-linker-TvRBL融合蛋白在大肠杆菌BL21(DE3)pLysS诱导表达的SDS-PAGE分析
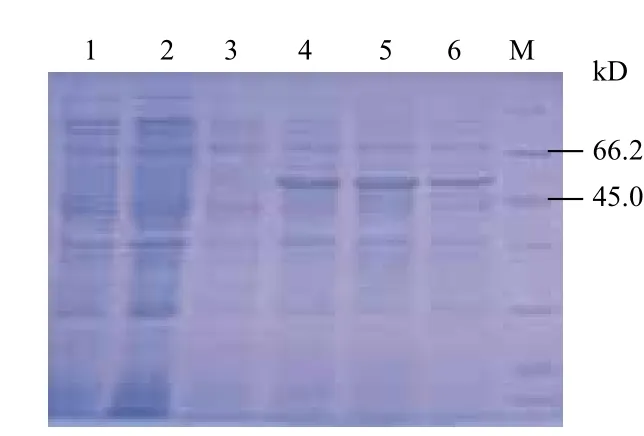
图10 ΔTvALP-linker-TvRBL融合蛋白在大肠杆菌BL21(DE3)pLysS诱导表达的SDS-PAGE分析
3 讨论
本研究通过基因重组,组建了TvALP-linker-TvRBL及ΔTvALP-linker-TvRBL融合蛋白。融合蛋白中的两个成分能否相互协同,发挥更好的生物活性,与二者能否分别形成正确的空间构象是密切相关的。目前,多数研究者采用低疏水性及低电荷效应的氨基酸组成的多肽接头充分伸展以分开两种组分,使它们能互不干扰地充分折叠成各自的天然构象。一般认为Linker的长度在5-25个氨基酸残基为适,太短不能提供足够的空间,较长对蛋白的折叠及稳定性有益,但容易受到蛋白酶的攻击。含有Gly-Ser的连接肽是目前报道使用较多的连接肽之一,其中甘氨酸是分子量最小、侧链最短的氨基酸,可增加侧链的柔性;丝氨酸是亲水性最强的氨基酸,可增加Linker的亲水性[12-14]。
钟丹丹等[15]通过添加10个中性氨基酸残基的柔性肽基因序列将CD59和CD2两类重要的淋巴细胞膜表面分子连接起来,成功构建了pIRES-CD59-linker-CD2融合基因真核表达系统。张明明等[16]添加19个中性氨基酸残基TS-(G4S)3-AC的Linker编码序列将心肌肌钙蛋白I(cTnI)和肌钙蛋白C(TnC)基因连接起来,成功构建了pET28a-cTnI-linker-TnC融合基因原核表达载体,并实现在大肠杆菌中可溶性表达。
为了保持融合蛋白的空间构型,本研究在两个基因之间添加了5个中性氨基酸的“小型柔性肽”,技术关键在于利用了linker基因中间的ggatcc序列恰好是BamHⅠ酶切位点这一特性,把linker基因分成两部分,分别放在TvALP和TvRBL基因的引物中,通过BamHⅠ酶切连接,成功拼接出了linker基因,筛选到了pET30a-TvALP-linker-TvRBL及pET30a-ΔTvALP-linker-TvRBL阳性重组质粒,并在E.coli BL21(DE3)pLysS宿主菌中得以表达,这与国内外一般采用合成单链,然后退火形成双链DNA实现连接,或直接合成DNA后再连接的技术路线相比较,具有更容易筛选到阳性重组子的优点。
pET30a系列载体是目前应用最为广泛的原核表达系统,已成功在大肠杆菌中表达了各种的异源蛋白[17,18]。本研究以pET30a质粒为表达载体,以E.coli BL21(DE3)pLysS为宿主表达菌,将TvALP-linker-TvRBL及切除信号肽的ΔTvALP-linker-TvRBL的融合基因分别克隆到原核表达载体,在相同条件下经IPTG诱导含重组质粒pET30a-TvALP-linker-TvRBL的菌株未诱导出蛋白,但含重组质粒pET30a-ΔTvALP-linker-TvRBL的菌株可以诱导融合蛋白的表达。原核生物翻译过程的正确起始于mRNA 5'端的SD序列及其SD序列与起始密码子之间的距离有关,30S亚基中16S rRNA必须与SD序列结合,才能使起始密码子正确进入P位点,并形成复合物。因此,mRNA 5'端的二级结构是影响翻译起始的一个重要因素,TvALP-linker-TvRBL信号肽的存在,使转录产物的5'端相对于去除信号肽的5'端形成复杂稳定的二级结构,从而影响到30S亚基与SD序列的识别与结合。
真核生物的信号肽在原核生物中进行表达时大部分不具有分泌功能,并且它始终与活性蛋白融合在一起,影响活性蛋白的正确折叠与功能发挥,需要在体外切除,因此,采用切除信号肽序列的ΔTvALP-linker-TvRBL基因在原核细胞中获得了表达。该研究需要进一步探究该融合蛋白的可溶性,为下一步融合蛋白的纯化,研究其抑制黄曲霉的分子作用机理奠定基础。
4 结论
本研究通过添加5个中性氨基酸的柔性多肽序列(linker)将TvRBL基因融合到TvALP基因的下游,成功构建了pET30a-TvALP-linker-TvRBL原核表达载体以及切除信号肽的pET30a-ΔTvALP-linker-TvRBL原核表达载体,切除信号肽后获得大小约60 kD的蛋白条带。与预测的目的产物蛋白条带大小一致。
[1]Szekeres A, Kredics L, Antal Z, et al. Isolation and characterization of protease overproducing mutants of Trichoderma harzianum[J]. FEMS Microbiology Letters, 2004, 233(2):215-222.
[2] Suárez MB, Vizcaíno JA, Llobell A, et al. Characterization of genes encoding novel peptidases in the biocontrol fungus Trichoderma harzianum CECT 2413 using the TrichoEST functional genomics approach[J]. Current Genetics, 2007, 51(5):331-342.
[3]纪明山, 李博强, 陈捷, 等.绿色木霉TR-8菌株对尖镰孢的拮抗机制[J].中国生物防治, 2005, 21(2):104-108.
[4]迟玉杰, 杨谦, 宋颖琦.对哈茨木霉转化子菌体形态观察及抗药性测定[J].哈尔滨工业大学学报, 2002, 34(6):784-788.
[5]Rodriguez-Kabana R, Kelley WD, Curl EA. Proteolytic activity of Trichoderma viride in mixed culture with Sclerotium rolfsii in soil[J]. Canadian Journal of Microbiology, 1978, 24(4):487-490.
[6]Guillot J, Konska G. Lectins in higher fungi[J]. Biochemical Systematics and Ecology, 1997, 25(3):203-230.
[7]Hori K, Ogata T, Kamiya H, et al. Lectin-like compounds and lectin receptors in marine microalgae:hemagglutination and reactivity with purified lectins[J]. Journal of Phycology, 1996, 32(5):783-790.
[8]Pemberton RT. Agglutinins(lectins)from some British higher fungi[J]. Mycological Research, 1994, 98(3):277-290.
[9]Mao HY. A self-cleavable sortase fusion for one-step purification of free recombinant proteins[J]. Protein Expres Purif, 2004, 37(1):253-263.
[10]Kim S, Lee YI. High-level expression and simple purification of recombinant human insulin-like growth factor I[J]. Journal of Biotechnology, 1996, 48(1):97-105.
[11]康彬, 童哲.一种利于蛋白质回收的快速SDS-聚丙烯酰胺凝胶电泳染色-脱色方法[J].生物化学与生物物理进展, 2000,27(2):210-211.
[12]Wen Q, Ma L, Luo W, et al. Expression, purification, and refolding of recombinant fusion protein hIL-2/mGM-CSF[J]. Biomed Environ Sci, 2008, 21:509-513.
[13]Sakamoto S, Taura F, Putalum W, et al. Construction and expression of specificity-improved single-chain variable fragments against the bioactive naphthoquinone, plumbagin[J]. Biol Pharn Bull, 2009,32:434-439.
[14]Yan RQ, Wu ZM, Fang QM, et al. Reconstruction of a chicken BF2 protein complex and identification of binding nonamer peptides derived from avian influenza virus hemagglutinin[J]. Vet Immunol Immunopathal, 2008, 126:91-101.
[15]钟丹丹, 高美华, 李伟伟, 等. CD59-linker-CD2 融合基因真核表达系统的构建[J].医学研究生学报, 2011, 24(1):15-19.
[16]张明明, 洪理泉, 卢仁泉, 等. cTnI-linker-TnC融合蛋白基因的构建, 表达及鉴定[J].现代检验医学杂志, 2007, 22(1):10-13.
[17]姜海霞, 刘永生, 杨孝朴, 等.人朊蛋白基因的克隆和原核表达[J].甘肃农业大学学报, 2006, 41(3):11-15.
[18]王晓东, 牟玉莲, 张莉, 等.近交五指山小型猪SLA-DQA基因的克隆及在大肠杆菌的原核表达[J].甘肃农业大学学报,2006, 41(4):23-26.
(责任编辑 马鑫)
Construction and Prokaryotic Expression of Trichoderma viride TvALP-linker-TvRBL Fusion Gene
Xu Yangyu1,2Wen Shijie1Li Haifen1Li Ling2Liang Xuanqiang1
(1. Crops Research Institute,Guangdong Academy of Agricultural Sciences,Guangzhou 510640;2. College of Life Science,South China Normal University,Guangzhou 510631)
Fusion gene were constructed by linking the TvALP and TvRBL genes with a flexible chain(linker). Bioinformatics analysis showed that the fusion gene contained a signal peptide. The TvRBL、TvALP and ΔTvALP genes were cloned by RT-PCR from the total RNA of Trichoderma viride mycelium, which in turn were cloned into expression plasmid pET30a to construct prokaryotic expression plasmids pET30a-TvALP-linker-TvRBL and pET30a-ΔTvALP-linker-TvRBL, then these two expression plasmids were transformed into E.coli BL21(DE3)pLysS, and protein expression were induced by IPTG. SDS-PAGE was used to analyze the expression of the fusion protein. The result showed that expression plasmid pET30a-ΔTvALP-linker-TvRBL was obtained expression in E.coli BL21(DE3)pLysS, which was a specific band at about 60 kD in size and identical with the expected molecular weight of the fusion protein.
Trichoderma viride;TvALP-linker-TvRBL fusion gene;signal peptide;prokaryotic expression
10.13560/j.cnki.biotech.bull.1985.2015.01.020
2014-05-22
国家“863”计划项目(2006AA10Z156),广东省自然科学基金重点项目(07117967)
徐杨玉,男,硕士,研究方向:生物防治微生物学;E-mail:574301612@qq.com
梁炫强,男,博士后,研究员,研究方向:花生遗传育种;E-mail:Liang804@yahoo.com
