全外显子组测序发现FUT6基因在1例糖尿病肾病中的终止突变
2015-10-22王玉华蔡春友李卫东韩鸿玲
王玉华,蔡春友,李卫东,韩鸿玲
(1.天津医科大学基础医学研究中心,天津300070;2.天津医科大学总医院肾脏科,天津300052)
随着人们生活水平的提高、饮食结构的改变和体力劳动的减少,糖尿病发病率与日俱增。根据国际糖尿病联盟(IDF)统计,2011年全球糖尿病患者数为3.66亿,预计2030年将达到5.52亿。糖尿病肾病(diabetic nephropathy,DN)是以糖代谢异常为主要原因所致的肾小球硬化并伴尿蛋白含量异常,是糖尿病的严重微血管并发症,亦是糖尿病患者的主要死亡原因之一,在我国糖尿病肾病已经成为导致终末期肾病的第二大原因,有资料表明每年新发的终末期肾病中DN占40%[1]。尽管以往研究认为糖尿病病程长和控制不良是DN的重要危险因素,但近来大量资料表明遗传因素在DN的发生发展中起关键作用[2-3],因此关于2型糖尿病肾病的遗传学研究成为近年来研究的热点。我们曾在1例无微量蛋白尿的早期DN患者的肾活检组织中,发现有TGF-β1、TGF-βR1 和 Smad3 表达升高,目前该患者已发展为终末期肾病。为研究该患者DN早期TGF-β途径激活的可能分子机制,进一步探寻与2型糖尿病肾病的相关致病基因,了解DN的遗传背景,我们对这例DN患者进行了全外显子测序。
1 资料与方法
1.1 研究对象 患者男性,年龄63岁,病程20年,1994年出现尿糖升高,行OGTT检查确诊为2型糖尿病,诊断标准参照2010年美国糖尿病协会公布的糖尿病诊断标准。2008年尿生化检查尿蛋白升高,诊断为DN(DN诊断标准为:有糖尿病病史且尿微量白蛋白/肌酐>300mg/g,除外原发性肾小球病和其他继发性肾小球疾病),在DN早期和中晚期两次行肾活检检查,病理表现均为糖尿病肾损害,确诊为糖尿病肾病,行眼底镜检查发现视网膜病变,同时伴有周围神经病变。自2005年起采用胰岛素治疗,目前该患者已发展为终末期肾病,采用血液透析治疗。本次研究调查和取样均征得受试者同意并签署了知情同意书。
1.2 实验方法
1.2.1 基因组DNA的提取 采用高盐法(北京百泰克生物技术公司,全血基因组DN提取试剂盒)提取外周血白细胞基因组DNA,应用Nanodrop2000/2000c分光光度计测定样品DNA浓度及OD值(A260/280,A260/230),记录样品浓度和纯度后-80℃储存备用。
1.2.2 全基因组外显子测序 采用AgilentSureSelect Human Kit芯片对DNA样品进行外显子捕获,样品外显子文库建立后通过Hiseq2000高通量双末端测序平台进行测序。输出的原始数据(Reads)用SOAPaligner2.20与参考基因序列(NCBIbuid36.3,hg18)比对,通过 SOAPsnp(v1.03)得到外显子及侧翼区域SNP位点数据。
1.2.3 候选基因筛选 首先筛选出非同义突变(non-synonymous,NS)和剪切位点(splice acceptor and donor,SS)突变,将这些可能引起功能改变的突变(NS/SS)和公共数据库 YH、dbSNP129、8HapMap外显子组、千人基因组计划进行逐步对比过滤,筛除已报道的基因多态性位点。由于无义突变对基因功能的影响更加明显,所以选出无义突变进行基因功能分析。
1.2.4 免疫组化 采用过氧化物酶标记的链霉卵白素(streptavidin/peroxidase,SP)法对患者肾活检组织切片进行免疫组织化学染色,研究基因FUT6(Abcam)的表达量。
2 结果
2.1 外显子测序结果 利用Hiseq2000新一代高通量双末端测序平台,对1例病理确诊的糖尿病肾病患者的全基因组外显子进行NS/SS突变分析,得到外显子及侧翼区域SNP数据库,见表1。
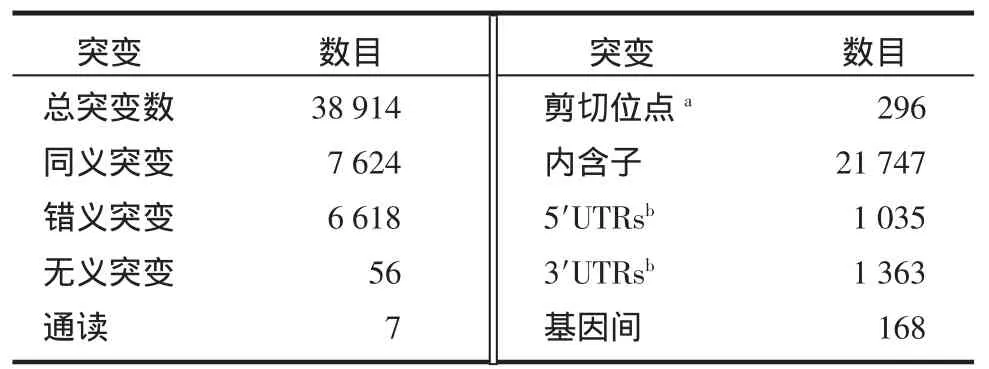
表1 样本外显子SNP检测结果Tab1 Summary of SNPs for exom e capture sample
2.2 外显子滤过结果 在测序结果中选出患者的NS和SS这些突变相比同义突变和非编码区突变对基因功能的影响更为显著。NS/SS突变经过公共数据库YH、dbSNP129、8HapMap外显子组和千人基因组外显子组数据逐步过滤,排除多态性,最后发现有509个突变不存在于这些数据库中(表2),其中包括497个错义突变和12个无义突变。对12个存在无义突变的基因(表3)在肾脏的表达情况进行生物信息学分析(genome.ucsc.edu),发现在肾脏高表达的基因有ANKRD35、ACSM2A、FUT6和HAAO。

表2 突变滤过过程和结果Tab2 Variant prioritization pipeline
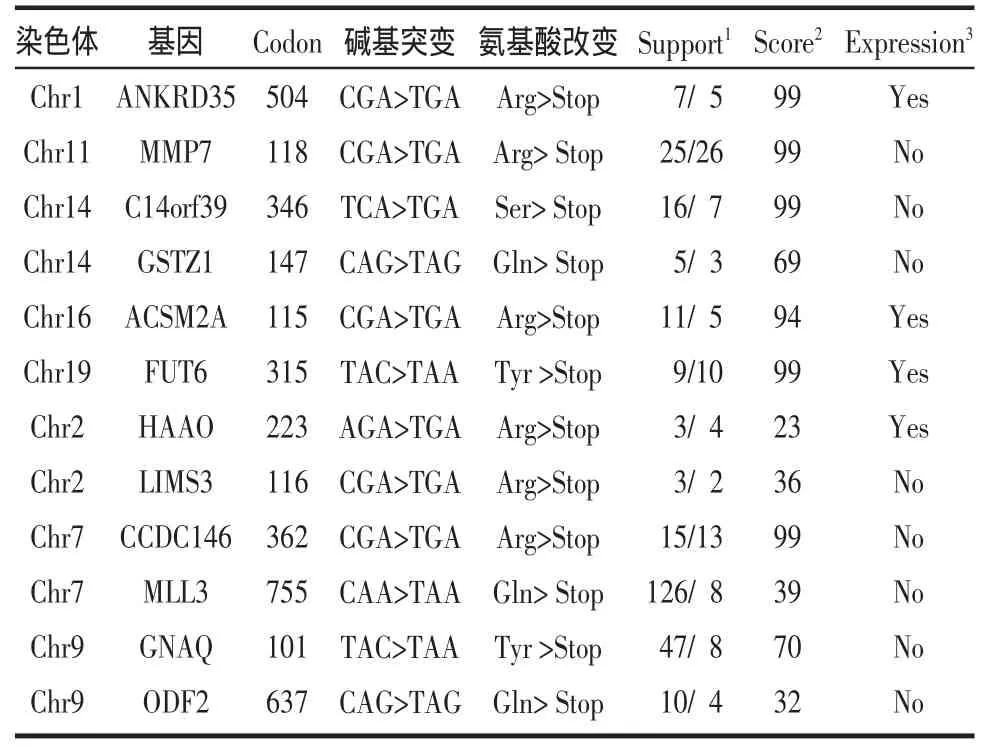
表3 样本无义突变概况Tab3 Summary of nonsensemutations
2.3 免疫组化结果 考虑突变位点的Support、Score、expression和基因功能分析,选取FUT6基因进行研究,免疫组化检测FUT6基因在该患者肾脏的表达,可以明显看出FUT6主要在肾小管表达,患者FUT6表达与正常人比较明显下降(图1)。

图1 DN患者与正常人肾组织FUT6表达水平(×400)Fig1 Theexpression levelofFUT6in DNpatientsandnorm alperso n(×400)
3 讨论
DN不仅是糖尿病患者的主要死亡原因,同样也是导致终末期肾病的主要原因之一,大量资料表明遗传因素在DN的发生和发展中起关键性作用。随着人类基因组计划(Human genome project,HGP)和人类基因组单体型图计划(the international haplotypemap project,HapMap)的相继完成,高通量测序技术的飞速发展,对个体全基因组或外显子序列进行测序,可获得完整的全基因组或外显子变异位点图谱,研究与疾病相关的基因。目前多项研究显示外显子测序在孟德尔疾病的研究中取得了重大突破,证实外显子测序发现孟德尔疾病的致病基因是有效和可行的[4-5]。
DN的确诊有赖于肾组织病理学检查,20世纪50年代以来,许多学者致力于DN病理结构的研究,Tervaert等[6]在2010年将T1DN和T2DN的组织学改变进行统一的病理分级,分别对肾小球、肾小管和肾血管损害进行评估,但国际仍未形成统一的病理分级。目前临床对DN的诊断和分级依据尿微量白蛋白,这与糖尿病合并肾脏疾病的患者很难区分,而糖尿病患者很少做肾活检,所以目前临床对DN的诊断并未十分明确。本研究以1例DN早期肾组织病理未见明显异常但出现TGF-β1、TGF-βR1和Smad3的表达升高,经临床和病理分析确诊的DN患者作为研究对象,对其进行全基因组外显子测序,以寻找与DN发病相关的致病基因,进一步较为全面的了解DN的遗传背景。
FUT6(岩藻糖基转移酶-6)基因定位于19p13.3,编码α-1,3-岩藻糖基转移酶的成员之一,催化岩藻糖基由GDP-α-L-Fuc转移至糖链外侧N-乙酰氨基葡萄糖(GlcNAc)3 位上的 FucT[7],其主要的功能是合成Lewis系抗原(Lewisa和Lewisx)和唾液酸化的Lewis系相关抗原(sLewisa和sLewisx),在肿瘤的发生和发展中起重要作用[8-9]。本研究中,患者FUT6基因的碱基发生315TAC>TAA突变,使得氨基酸残基由酪氨酸突变为终止密码子,翻译提前终止。Mollicone[10]在研究血清缺乏α-1,3-岩藻糖基转移酶的印尼人家系时也发现了该突变位点的存在,证明了FUT6基因发生315Try>stop突变时,血清α-1,3-岩藻糖基转移酶缺乏,从而影响其功能。有研究报道缺乏糖基化修饰的TGF-βR2几乎不与TGF-β1结合[11],具体的调节机制并不是十分明确,但Kim[12]用免疫荧光的方法证明TGF-βR2的糖基化修饰影响其在细胞表面的定位,从而影响与TGF-β1的结合,进而影响其功能,这说明TGF-βR2的糖基化修饰是TGF-β1致纤维化的必要条件。Hirakawa[13]在研究 α-1,3F UTs对结肠癌细胞的生物学行为的影响时发现,用siRNA干扰FUT6和FUT3后TGF-βR1和TGF-βR2的岩藻糖基化修饰明显减少,Western blot发现 E-cadherin、pHSP27 和 pP38的表达明显下降,曾有报道E-cadherin[14]和pP38[15]在TGF-β介导的上皮间充质转化(epithelialmesenchymal transition,EMT)中有重要作用,EMT 是肾脏纤维化的重要过程[16],由此可以认为FUT6对TGF-βRs的岩藻糖基化修饰可能在肾间质纤维化过程中起到重要作用。
目前没有发现DN遵循一种明确的孟德尔遗传模式,认为是多种遗传因素和环境因素共同作用的结果。本次研究对一早期还未出现肾脏功能性改变,但免疫组化证实有TGF-β、TGF-βR1和Smad3高表达的DN患者的全外显子进行测序,结合基因功能学分析发现基因FUT6的终止突变,免疫组化证实在该患者的肾活检标本中FUT6的表达下降。但目前尚缺乏直接证据表明FUT6终止突变与早期出现的TGF-β/Smad信号通路的激活有直接的联系,需要进一步分子生物学实验对其致病的具体机制进行研究。另外候选致病基因还需进行相关病例疾病验证和种系突变检测,以明确其在该疾病中的发生率。
[1]Kikkawa R,Koya D,HanedaM.Progression ofdiabetic nephropathy[J].AJKD,2003,41(3Suppl1):S19
[2]Arar NH,Freedman B I,Adler SG,etal.Heritability of the severity of diabetic retinopathy:The FIND-Eye study[J].Invest Ophthalmol Vis Sci,2008,49(9):3839
[3]Wong TY,Klein R,Islam A,etal.Diabetic retinopathy in amultiethnic cohort in the United States[J].Am JOphthalmol,2006,141(3):446
[4]Ng SB,Turner E H,Robertson P D,etal.Targeted capture and massively parallelsequencingof12 human exomes[J].Nature,2009,461(7261):272
[5]Walsh T,Shahin H,Elkan-Miller T,etal.Wholeexome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82[J].Am JHum Genet,2010,87(1):90
[6]Tervaert TW,Mooyaart A L,Amann K.Pathologic classification of diabetic nephropathy[J].JAm Soc Nephrol,2010,21(4):556
[7]Sherwood A L,Upchurch D,Stroud M R,etal.A highly conserved His-His motif present in alpha1-->3/4fucosyltransferases is required for optimal activity and functions in acceptor binding[J].Glycobiology,2002,12(10):599
[8]Holgersson J,Lofling J.Glycosyltransferasesinvolved in type1 chain and Lewis antigen biosynthesis exhibit glycan and core chain specificity[J].Glycobiology,2006,16(7):584
[9]Dyugovskaya L,Lavie P,Lavie L.Increased adhesion molecules expression and production of reactive Oxygen species in leukocytes ofsleep apnea patients[J].Am JRespirCritCareMed,2002,165(7):934
[10]Mollicone R,Reguigne I,Fletcher A,etal.Molecular basis for plasma alpha(1,3)-fucosyltransferase gene deficiency(FUT6)[J].J BiolChem,1994,269(17):12662
[11]Wang X,Inoue S,Gu J,etal.Dysregulation of TGF-beta1 receptor activation leads toabnormal lung developmentand emphysema-like phenotype in core fucose-deficientmice[J].PNAS,2005,102(44):15791
[12]Kim YW,Park J,Lee H J,etal.TGF-beta sensitivity isdetermined by N-linked glycosylation of the type II TGF-beta receptor[J].Biochem J,2012,445(3):403
[13]Hirakawa M,Takimoto R,Tamura F,etal.Fucosylated TGF-beta receptors transducesa signal for epithelial-mesenchymal transition in colorectalCancer cells[J].Br JCancer,2014,110(1):156
[14]Ge SA,Costa R S,Ravinal R C,etal.Mast cells,TGF-beta1 and alpha-SMA expression in IgA nephropathy[J].Dis Markers,2008,24(3):181
[15]Yu L,HebertM C,Zhang Y.TGF-beta receptor-activated p38MAP kinasemediates Smad-independent TGF-beta responses[J].EMBO,2002,21(14):3749
[16]Zhao C G,He X J,Lu B,etal.Increased expression of collagens,transforming growth factor-beta 1,and-beta 3 in glutealmuscle contracture[J].BMCMusculoskeletDisord,2010,11:15
