高效液相色谱法测定恩替卡韦口服液中有关物质
2015-10-18严拯宇廖声华
严拯宇, 黄 玉, 王 瑞, 廖声华
(中国药科大学理学院,药物质量安全与预警教育部重点实验室,江苏南京 211198)
恩替卡韦(2-氨基-9-[(1S,3R,4S)-4-羟基-3-羟甲基-2-亚甲基环戊基]-1,9-二氢-6H-嘌呤-6-酮-水合物)是一种鸟嘌呤核苷类似物,能够有效和选择性地对抗乙肝病毒,比拉米夫定和阿德福韦能更快、更有效地抑制乙肝病毒的复制[1]。目前已有恩替卡韦原料药含量及有关物质测定[2,3]、恩替卡韦胶囊的含量测定[4]、恩替卡韦分散片及其有关物质测定[5-7]等报道。但口服液剂型的恩替卡韦有关物质的研究未见报道。由于口服液辅料和工艺的改变,可能会带入一些杂质,而药品中的杂质成分直接关系到药品的安全性问题,因此控制药品中杂质含量是至关重要的。恩替卡韦原料药中主要含有3种非对映异构体,即杂质A(2-氨基-9-[(1R,3R,4S)-4-羟基-3-羟甲基-2-亚甲基环戊基]-1,9-二氢-6H-嘌呤-6-酮)、杂质B(2-氨基-9-[(1R,3R,4R)-4-羟基-3-羟甲基-2-亚甲基环戊基]-1,9-二氢-6H-嘌呤-6-酮)、杂质C(2-氨基-9-[(1S,3R,4R)-4-羟基-3-羟甲基-2-亚甲基环戊基]-1,9-二氢-6H-嘌呤-6-酮)。主成分及杂质的结构式见图1。
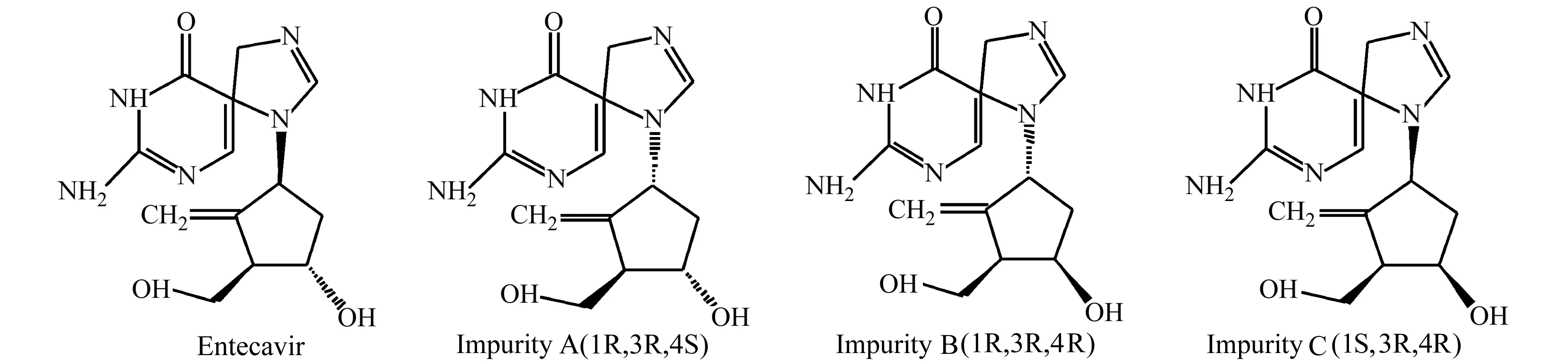
图1 恩替卡韦及杂质A、B和C的结构式
本实验参照国家药品标准,建立了准确、灵敏的梯度洗脱方法[8],采用加校正因子的主成分自身对照法计算杂质的含量,保证制剂的安全性。
1 实验部分
1.1 仪器与试剂
LC-20AT高效液相色谱仪(日本,岛津公司),配有SPD-M20A二极管阵列检测器(DAD);Agilent ZORBAX SB-C18色谱柱(150×4.6 mm,3.5 μm)(美国,Agilent);BT25S型电子天平(赛多利斯)。
恩替卡韦(93.7%,ID:NL8U-DR63)由中国食品药品检定研究院提供,用于含量测定。杂质A对照品(96.3%,批号:20140415)、杂质B对照品(97.7%,批号:20140425)、杂质C对照品(99.6%,批号:20140410),均购于海门慧聚药业有限公司。乙腈(色谱纯,德国Merk公司),三氟乙酸(分析纯,南京化学试剂有限公司)。
恩替卡韦原料药、口服液(规格:50 μg/mL,批号:131022、131023、131024)及空白辅料由江苏汉晨药业提供。市售口服液(规格:50 μg/mL,批号: 3E76102)购自百时美施贵宝公司。
1.2 溶液制备
1.2.1对照品溶液取恩替卡韦对照品10.0 mg,精密称定,置于100 mL容量瓶中,加水超声溶解后稀释至刻度,移取5.00 mL于10 mL容量瓶中,加水定容、即得恩替卡韦对照品溶液。分别取杂质A、B、C对照品各1.00 mg,精密称定,置于50 mL容量瓶中,配制单个杂质储备液,移取0.125 mL,置于10 mL容量瓶中,加水定容、分别得杂质A、B、C对照品溶液。再精密移取各储备液适量,混合均匀,得系统适用性溶液(含主成分50.0 μg/mL和杂质A、B、C各0.25 μg/mL)。
1.2.2供试品溶液口服液和空白辅料经滤头(0.45 μm)过滤后,直接进样测定。
1.3 色谱条件
色谱柱为Agilent ZORBAX SB-C18柱;以体积比为97∶3∶0.15的水-乙腈-三氟乙酸为流动相A,乙腈为流动相B;梯度洗脱条件:0~3.5 min,100%A;3.5~25 min,100%~94.0%A;25~26 min,94.0%~89.2%A;26~28 min,89.2%~76.0%A;28~48 min,76.0%~70.0%A;48~50 min,70.0%~100%A;50~60 min,100%A;检测波长:254 nm;流速:1.0 mL/min;柱温:30 ℃;进样量:20 μL。
2 结果与讨论
2.1 色谱分离条件考察
取系统适用性溶液、口服液和空白辅料各20 μL注入高效液相色谱仪,记录色谱图(图2)。系统适用性色谱图中,主峰与相邻杂质峰的分离度RS为2.557,理论板数N以恩替卡韦峰计为21 832,分离效果与峰对称性均较理想,能满足有关物质测定的要求。
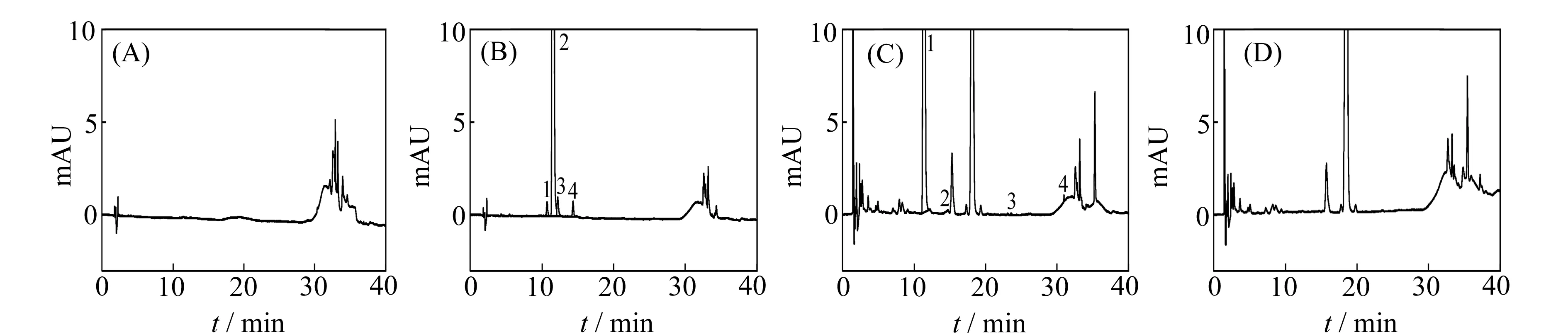
图2 系统适用性溶液的HPLC图
2.1.1流动相的选择参照文献方法[8],选用25 mmol/L磷酸盐缓冲液为流动相A,乙腈为流动相B,记录的色谱图显示较大的梯度干扰峰,改变进样梯度、更换新的流动相、色谱柱并冲洗管路后,梯度峰仍然存在。根据恩替卡韦片国家药品标准[9],选用水-乙腈-三氟乙酸为流动相A并调节体积比和洗脱程序,当三者体积比为97∶3∶0.15时,主成分与杂质B的分离度达2.0以上,符合系统适用性试验要求。
2.1.2检测波长的选择取系统适用性溶液进样,在波长190~400 nm范围内进行扫描。结果表明恩替卡韦和杂质A、B、C在254 nm处均有较大吸收,故选择254 nm作为检测波长。
2.1.3溶液浓度的选择由于口服液本身浓度仅50.0 μg/mL,故口服液不经稀释直接作为供试液。若自身稀释对照浓度为0.1%,则主成分峰面积太小,因此选择自身稀释对照溶液为0.25 μg/mL。
2.2 检测限和定量限
取系统适用性溶液,按稀释法测得杂质A、B和C的检测限分别为7.22、7.30、7.50 ng/mL,分别相当于恩替卡韦浓度的0.014%、0.015%、0.015%;杂质A、杂质B、杂质C的定量限分别为24.1、24.3、25.0 ng/mL,分别相当于恩替卡韦浓度的0.048%、0.049%、0.050%。
2.3 标准曲线
以水为溶剂配制含恩替卡韦主成分和杂质A、B、C各2.50 μg/mL的混合储备液,再用水稀释分别配制10%、50%、75%、100%、125%、150%限度浓度的标准系列溶液。以浓度(X,μg/mL)为横坐标,以色谱峰面积(Y)为纵坐标绘制标准曲线,杂质A、B、C及恩替卡韦在24.08~361.0、24.33~364.9、25.00~375.0、23.40~351.0 ng/mL范围呈良好线性。回归方程、相关系数和相对校正因子见表1。
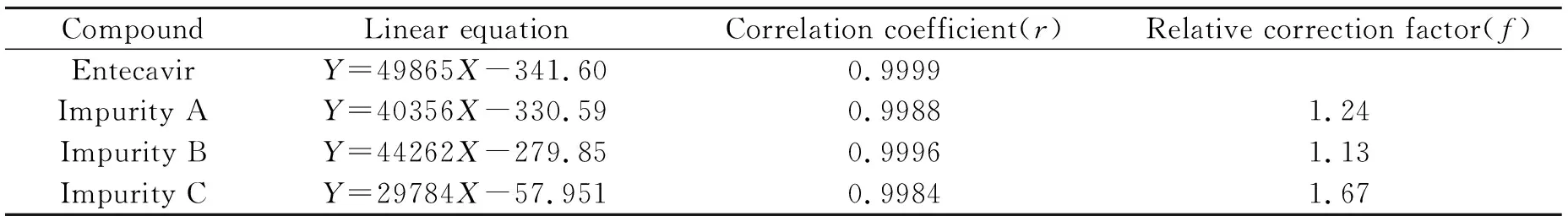
表1 回归方程、相关系数和相对校正因子
2.4 精密度试验
取100%标准系列溶液连续进样6次,测得杂质A、B、C的峰面积的相对标准偏差(RSD)分别为0.62%、2.5%和2.6%,精密度良好,符合有关物质测定的要求。
2.5 重复性试验
取6份口服液(批号:131023),分别精密移取口服液各0.500 mL于100 mL容量瓶中,用水稀释至刻度,摇匀,为0.5%自身稀释对照溶液。取各口服液和0.5%对照溶液进行重复性考察。结果口服液中未检测出杂质B和C;杂质A、最大单个杂质和总杂质的平均百分含量分别为0.04%(RSD=14%)、0.11%(RSD=11%)、0.23%(RSD=9.0%),表明此方法重复性良好。
2.6 稳定性试验
精密吸取单个杂质储备液适量,配制杂质对照溶液(分别含杂质A、B、C各0.25 μg/mL),与口服液分别于0、2、4、6、8、10、12、24 h进行色谱分析。杂质对照溶液中,杂质A、杂质B、杂质C峰面积的RSD(n=8)分别为3.5%、1.5%、2.2%,口服液中未检测到杂质B和C,杂质A、最大单个杂质和总杂质峰面积的RSD(n=8)分别为9.2%、7.8%和8.0%,表明杂质对照溶液和口服液在24 h内稳定性良好。
2.7 回收率试验
精密吸取混合储备液适量,配制回收率储备液(含恩替卡韦主成分和杂质A、B、C各5.00 μg/mL)。再用水稀释,分别配制50%、100%、150%的回收率测定供试液,每个浓度各三份。同2.6中的方法,配制50%、100%、150%的自身稀释对照液。回收率结果见表2,3个添加水平下杂质A、B、C的回收率均在70%~130%之间,满足有关物质的检测要求。
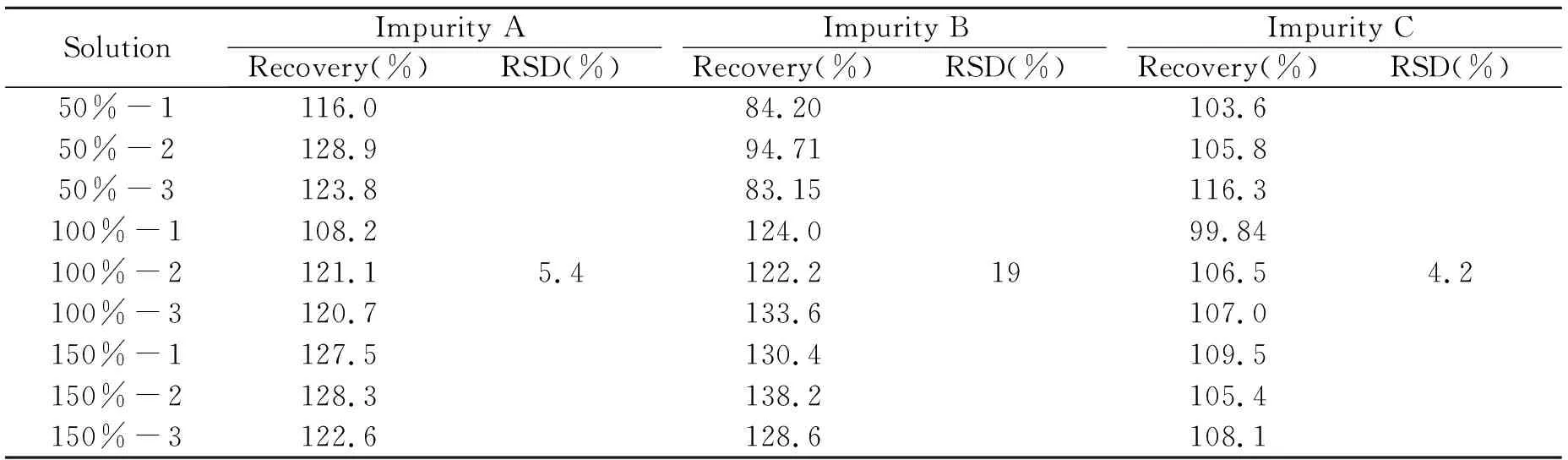
表2 杂质A、B、C的回收率结果
2.8 耐用性试验
改变系统参数如流动相A中水-乙腈-三氟乙酸体积比(97∶3∶0.10、97∶3∶0.15、97∶3∶0.20)、流速(±0.1 mL/min)和柱温(±2 ℃)等,各成分的保留时间会略有改变,但主峰不受相邻杂峰的影响。另外,该系统对色谱柱要求较苛刻,由于主成分恩替卡韦和杂质B难以分离,适当添加三氟乙酸能改善峰形,减少拖尾。但由于三氟乙酸浓度为0.15%,流动相的pH在2.0以下,因此对于色谱柱的pH耐受性要求较高。色谱柱选用Agilent ZORBAX SB-C18时,主成分恩替卡韦的峰和辅料峰、有关物质峰均能同时测定。
2.9 峰纯度检查
分别取恩替卡韦口服液、空白辅料及原料药适量,分别经酸破坏(0.1 mol/L HCl,50 ℃水浴,2 h)、碱破坏(0.1 mol/L NaOH,60 ℃水浴,80 min)、氧化破坏(10% H2O2,80 ℃水浴,1 h)、光破坏(强光照射24 h)、热破坏(沸水浴加热1 h)处理,其中酸破坏和碱破坏先加碱或酸中和至中性,分别用水稀释成含恩替卡韦浓度约为25 μg/mL的破坏供试液。另取口服液、空白辅料及原料药适量,用水溶解并稀释成未破坏供试液,进行HPLC-DAD测定(图3)。在所建立的色谱系统之下,恩替卡韦的色谱峰与分解、降解产物的分离良好;用DAD检测器对恩替卡韦主峰的峰纯度作出进一步判断,恩替卡韦的色谱峰是纯的。
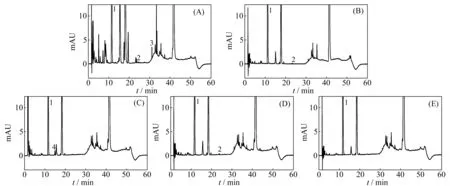
图3 恩替卡韦口服液强降解试验HPLC图
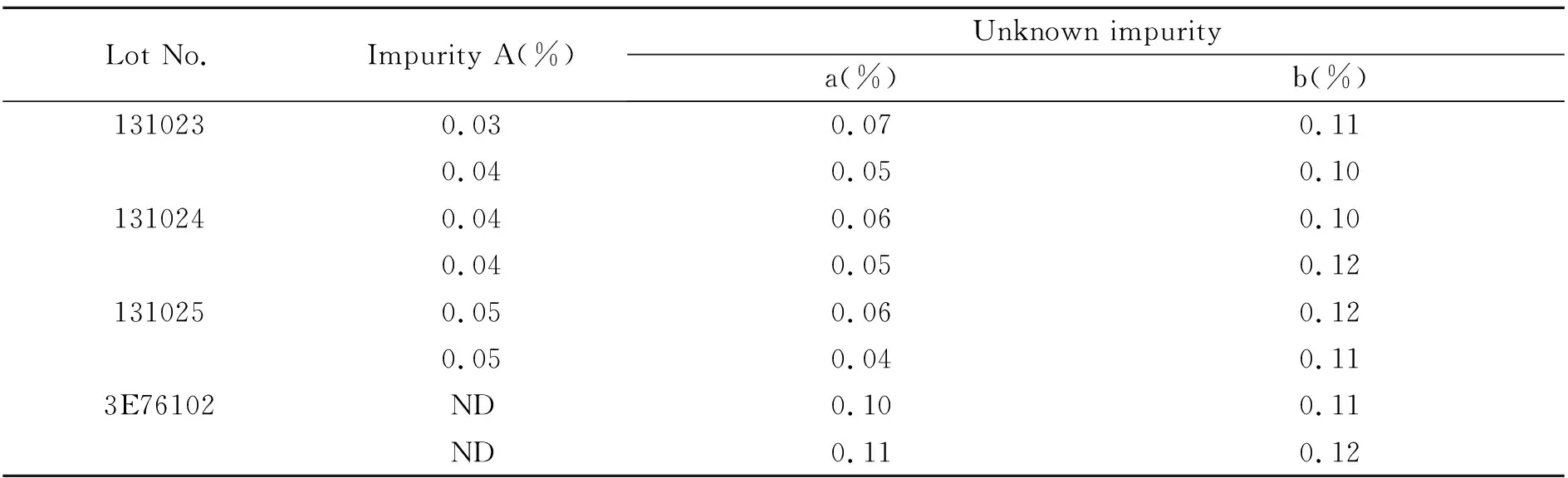
表3 样品含量测定结果(n=2)
2.10 含量测定
采用0.5%自身对照法分别对三批和一批市售口服液进行有关物质检查,结果见表3。三批口服液中均未检出杂质B和C,批号131023、131024、131025中杂质A的含量分别为0.04%、0.04%和0.05%,最大单个杂质的含量分别为0.1%、0.1%和0.1%,总杂质含量分别为0.2%、0.2%和0.3%;市售口服液中未检出杂质A、B和C,最大单个杂质的含量为0.1%,总杂质含量为0.2%。将单个杂质的限度暂定为0.2%,总杂质的限度定为0.5%。
3 结论
本研究建立了用HPLC-DAD测定恩替卡韦口服液中有关物质的分析方法,该方法以加校正因子的主成分自身对照定量,精密度高、重复性好,可用于恩替卡韦口服液中有关物质的测定。
