混合胶束毛细管电动色谱法测定片仔癀中有效成分
2015-10-18余丽双丛日琳林诗瑶孙照霞褚克丹
余丽双, 丛日琳, 林诗瑶,孙照霞,李 煌,林 埔,褚克丹,徐 伟
(1.福建中医药大学药学院,福建福州 350122;2.荣成市中医院,山东威海 264300)
片仔癀(Pien Tze Huang)是我国的传统名贵中成药,主要由牛黄、三七、蛇胆、麝香等多种名贵中药精制而成,具有清热解毒,凉血化瘀,消肿止痛之功效,可以消除人体内的“湿、热、毒、邪”,达到祛邪安正、预防保健的效果。对于片仔癀的质量控制,原标准收载于卫生部标准中药成方制剂第十八册只有两个反应及三七的薄层色谱鉴别项目[1]。《药典》记载了三七皂苷R1、人参皂苷Rg1、人参皂苷Rb1、胆酸、去氧胆酸的薄层色谱鉴别,对麝香酮制定了气相色谱的含量测定标准。为了更快速、准确地控制片仔癀的质量,有必要建立片仔癀的多成分含量测定的分析方法。
目前有关片仔癀多成分含量测定的报道仅见液相色谱(LC)法[2 - 4],尚未见应用毛细管电泳(CE)法分离检测的报道。本实验采用的CE法具有分析时间短、进样量小、毛细管柱便宜且易清洗、分析成本低等优点,更值得关注的是其分离模式多,且易于变换分离分析条件,相比于LC等技术,CE是罕见的适用于复杂样品体系及临床检验的分析手段,因此CE可为复杂中药及其制剂成分测定及质量控制提供有效的新方法[5 - 8]。本文采用混合胶束毛细管电动色谱(mixMs-MEKC)法分离检测片仔癀中三七皂苷R1、人参皂苷Rg1、麝香草酚、人参皂苷Rb1及牛磺胆酸钠5种有效成分,为片仔癀的质量监控和药理研究提供一种快速、灵敏而低成本的新的分析手段。
1 实验部分
1.1 仪器与试剂
P/ACETMMDQ型毛细管电泳仪(美国,Beckman公司),配备二极管阵列检测器(DAD)和32 Karat 5.0工作站;未涂层弹性石英毛细管柱(美国,Beckman公司,63.6 cm×75 μm i.d,有效长度52.0 cm);雷磁pHS-3C型酸度计(上海精密科学仪器有限公司);HH-4型数显恒温水浴锅(国华电器有限公司);XS105型电子分析天平(梅特勒-托利多国际股份有限公司)。
三七皂苷R1、麝香草酚及牛磺胆酸钠标准品(中国药品生物制品检定所);人参皂苷Rg1、人参皂苷Rb1标准品(成都曼斯特生物科技有限公司);十二烷基硫酸钠(SDS)和胆酸钠(SC)(北京百灵威科技有限公司);乙醇、乙腈、Na2B4O7等试剂为分析纯(国药集团化学试剂有限公司)。实验用水由Mili-Q超纯水仪制备。
片仔癀片剂(漳州片仔癀药业股份有限公司)。
1.2 实验方法
精密称取三七皂苷R1、人参皂苷Rg1、麝香草酚、人参皂苷Rb1及牛磺胆酸钠标准品各1.0 mg,用甲醇溶解,配成浓度为1.0 mg/mL的标准对照品溶液,使用时用超纯水稀释成所需浓度即可。将片仔癀片剂研磨成粉,精密称定干燥粉末0.5 g,用20 mL无水乙醇超声提取30 min,静置过滤,残渣重复提取一次,合并两次滤液,用氮吹仪浓缩至4.0 mL左右,无水乙醇溶解并定容至5 mL。进样前需经0.22 μm微孔滤膜过滤。
新毛细管分别用超纯水、0.1 mol/L的HCl 、超纯水、0.1 mol/L NaOH溶液、超纯水各冲洗20 min。使用过的毛细管分别以超纯水、0.1 mol/L NaOH溶液、超纯水和运行缓冲液各冲洗5 min即可,两次进样间用缓冲液冲洗2 min即可。所有溶液在电泳前均用聚乙烯滤膜(0.22 μm)过滤以去除大颗粒杂质,并经超声除去气泡。
2 结果与讨论
2.1 电泳分离条件的优化
2.1.1运行缓冲液及其pH值的优化实验先采用10 mmol/L Na2B4O7-20 mmol/L NaH2PO4作为缓冲运行液,考察不同pH值(8.0、8.5、9.0、9.5、10.0)对5种目标化合物的分离效果。结果发现pH为9.0和9.5时的分离效果均较好。考虑到配制缓冲溶液的方便性,最终选择可以达到同样分离效果的10 mmol/L的Na2B4O7溶液(pH≈9.2)为运行缓冲液。
2.1.2表面活性剂的种类及浓度的优化当表面活性剂的浓度高于胶束临界浓度时,易在水中形成胶束,产生类似色谱固定相的作用。所以表面活性剂的种类和浓度是MEKC条件选择的关键之一。实验考察了不同表面活性剂种类和浓度,如SC(70、75、80、85、90 mmol/L)、SDS(20、25、30、50 mmol/L)等,对5种化合物分离的影响,结果表明采用单一胶束均无法完全分离上述5种化合物。
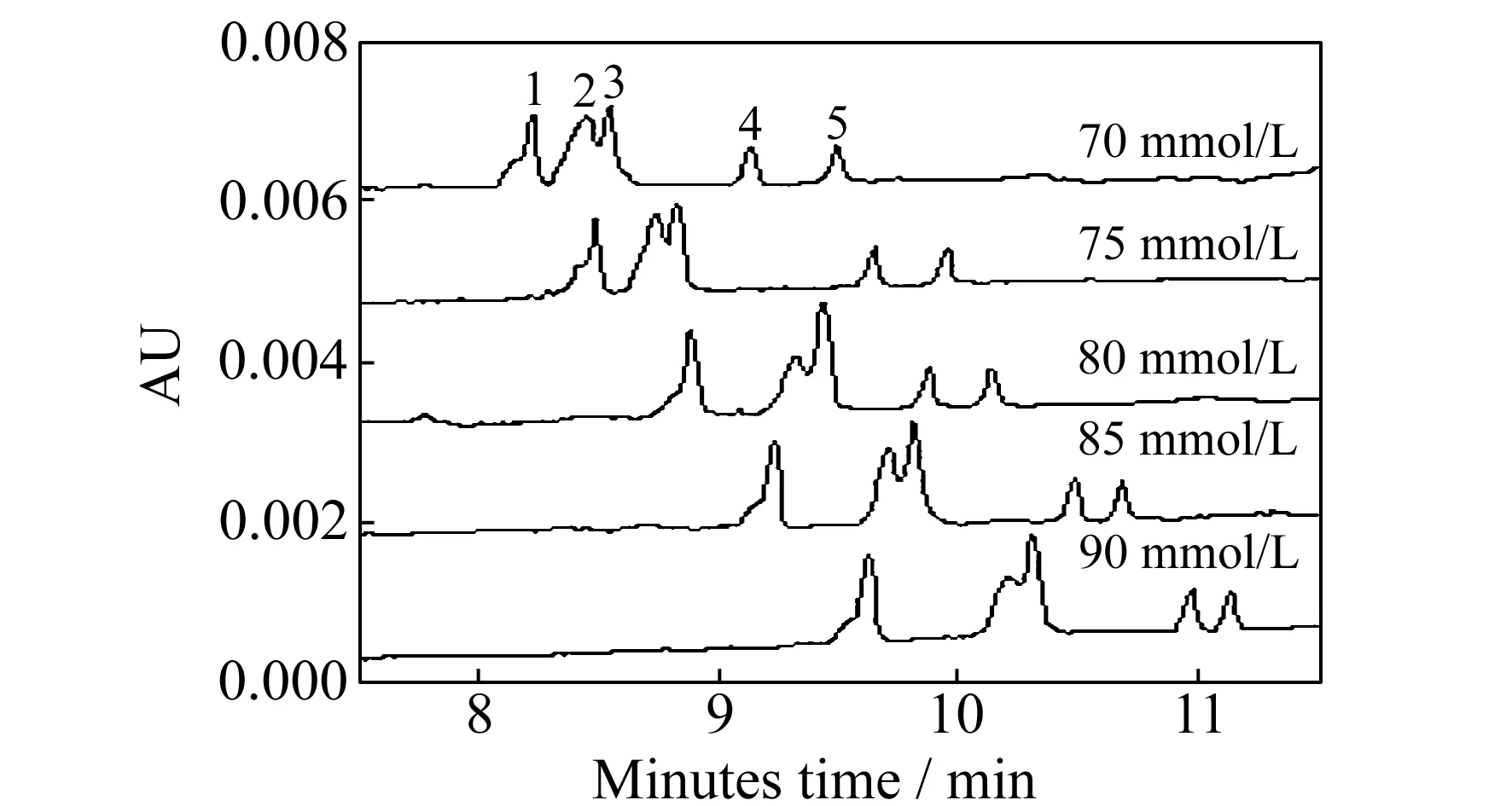
图1 胆酸钠(SC)浓度对5种化合物分离的影响
实验考察了混合胶束对分离的影响,发现采用SC-SDS二元混合胶束可以有效分离片仔癀中的5种化合物。实验首先优化了SC浓度(70、75、80、85、90 mmol/L)对分离产生的影响。结果表明,随着SC浓度的增大,分析物的迁移时间延长,分离度逐渐增大;当SC浓度大于85 mmol/L时,人参皂苷Rg1(峰2)和麝香草酚(峰3)的分离度明显减小,因此,选择SC的最优浓度为85 mmol/L,如图1。在上述最优化SC单一胶束溶液中加入SDS形成二元混合胶束,进一步考察SDS浓度(20、25、30、50 mmol/L)对分离的影响,发现随着SDS浓度增加,迁移时间延长,分离改善,当SDS浓度为30 mmol/L时,即当运行缓冲液为10 mmol/L Na2B4O7+85 mmol/L SC+30 mmol/L SDS(pH≈9.2)时,5种化合物均得到基线分离。本实验发现采用混合二元胶束分离效果优于单一胶束其可能的原理如下:(1)混合二元胶束的增溶效果较单一胶束好,化合物与混合胶束可相互作用的范围增大,即其分离窗口增大,可能使分离得到改善;(2)各分析物在水相与假固定相混合胶束之间的分配系数发生变化,可能改善其分离效果。
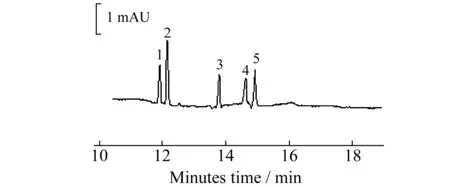
图2 混合标准品的MEKC分离图
2.1.3分离电压及进样时间的优化随着分离电压的增大,分离效率增大,但当电压过高时,会产生较多的焦耳热,引起区带展宽,使分离效率下降。综合考虑分离度、分析时间及峰展宽等因素,选择25 kV为最佳分离电压。对进样时间进行优化,实验发现当进样时间大于10 s(0.5 psi)时,峰形展宽,故选择10 s(0.5 psi)为最佳进样时间。
综上所述,在运行缓冲液为10 mmol/L Na2B4O7+85 mmol/L SC+30 mmol/L SDS(pH≈9.2),进样时间为10 s(0.5 psi),分离电压为25 kV的最佳条件下,片仔癀中5种有效成分标准混合液的电泳图见图 2。5种物质在16 min内达到基线分离。
2.2 方法学考察
2.2.1线性关系及检测限在上述最优条件下,测定5种化合物在一定浓度范围内的峰面积和迁移时间,以各成分的浓度(X,μg/mL)对其峰面积(Y)进行线性回归。根据三倍信噪比(S/N=3)测定各组分的检测限。结果如表1 所示,5种化合物的线性关系良好。
2.2.2精密度试验在上述最优条件下,取一定浓度的混合标准品溶液,连续进样5次,计算各组分的峰面积和迁移时间,并计算相对标准偏差(RSD),结果显示该方法的精密度良好,结果见表1。
2.2.3稳定性试验取一定浓度的各组分对照品溶液,分别于0、2、16、18、24 h考察各组分峰面积的变化(用RSD表示)。结果表明,除麝香草酚外各组分在24 h内较为稳定,三七皂苷R1为3.9%,人参皂苷Rg1为4.77%,人参皂苷Rb1为3.17%。麝香草酚在16 h后稳定性较差(RSD>10%),溶液需要重新配制。
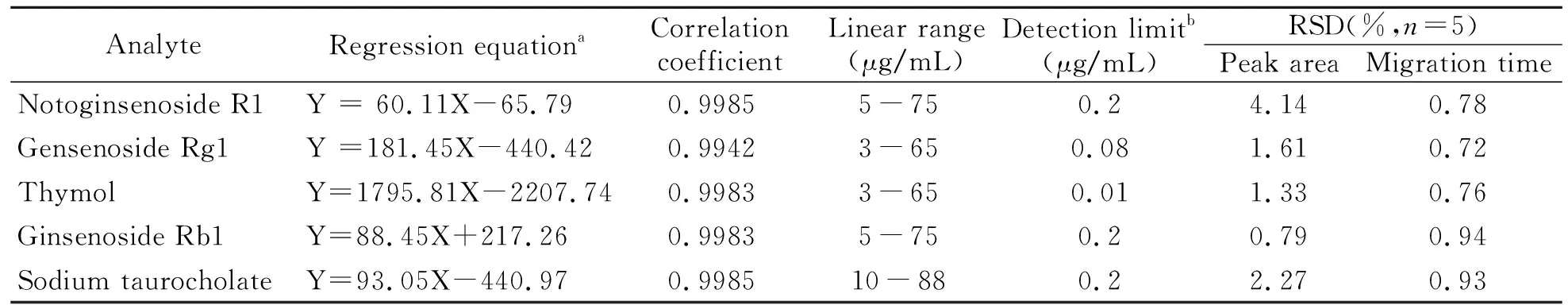
表1 回归方程、线性范围、检测限和精密度
aWhere theYandXare the peak area and concentration of the analytes(μg/mL),respectively;bThe detection limits corresponding to concentrations giving signal to noise ratio of 3.
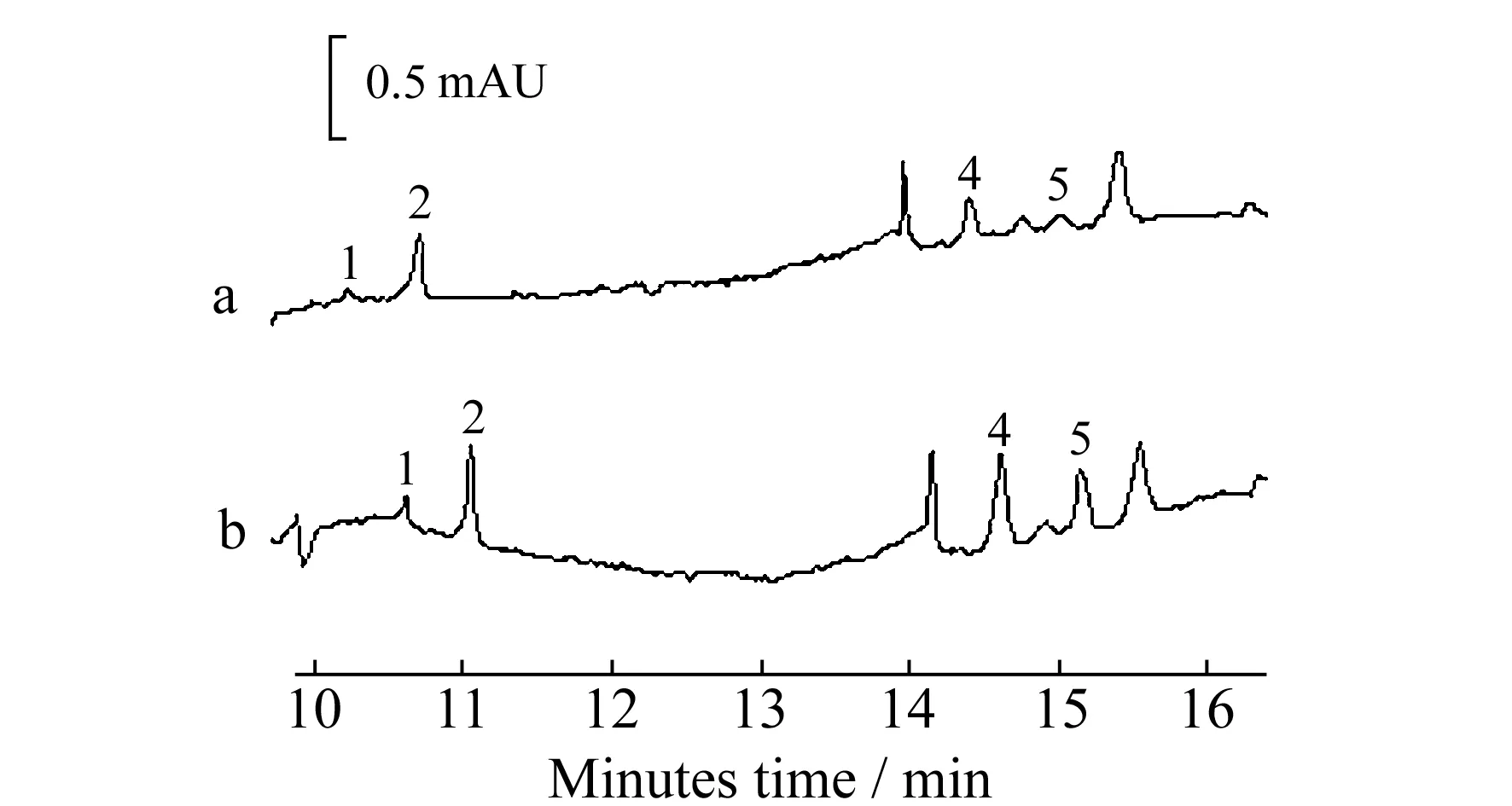
图3 片仔癀样品的电泳色谱图
2.3 实际样品含量测定及回收率试验
在最优条件下,采用标准加入法测定供试品溶液,其电泳谱图如图 3所示。由图3可见,除了麝香草酚由于不稳定提取过程损失,其他4种成分均可进行定性分析,通过外标法测定片仔癀中各有效成分的含量见表 2。
精密称定片仔癀样品粉末,按实验方法制备供试品溶液,然后取一定浓度的供试品溶液,加入适量的标准品溶液,进行加标回收率试验,结果见表2。由表2 可见,除了牛黄胆酸钠由于其共存杂质的影响,无法测定回收率,其他的回收率在76.1%~112.8%之间,基本上满足了分析方法的要求,有望为片仔癀的质量控制提供有效的新方法。
3 结论
本实验建立了一种同时测定片仔癀中的三七皂苷R1、人参皂苷Rg1、麝香草酚、人参皂苷Rb1及牛磺胆酸钠等5种化合物的混合胶束毛细管电动色谱新方法,实验发现混合二元胶束的协同作用可以改善分离效果。结果表明所建新方法简单快速,其线性关系良好,精密度高,有望为片仔癀的质量控制和药理研究提供有效的新方法。

表2 片仔癀样品的回收率及含量测定
aNS:refers to not separated.
