非综合征型遗传性耳聋家系的遗传学特征分析
2015-09-18牛志杰孙捷梅凌云蒋璐陈红胜贺楚峰刘亚兰王雪萍文杰熊俊冯永11中南大学湘雅医院耳鼻咽喉头颈外科长沙4100082耳鼻咽喉重大疾病研究湖南省重点实验室长沙410008中南大学医学遗传学国家重点实验室长沙4100784新疆医科大学第一附属医院耳鼻咽喉科乌鲁木齐80011
牛志杰 孙捷 梅凌云 蒋璐 陈红胜 贺楚峰 刘亚兰 王雪萍 文杰 熊俊 冯永1,2,1中南大学湘雅医院耳鼻咽喉头颈外科(长沙410008)2耳鼻咽喉重大疾病研究湖南省重点实验室(长沙410008)中南大学医学遗传学国家重点实验室(长沙410078)4新疆医科大学第一附属医院耳鼻咽喉科(乌鲁木齐80011)
·临床研究·
非综合征型遗传性耳聋家系的遗传学特征分析
牛志杰12孙捷1,2,4梅凌云12蒋璐12陈红胜12贺楚峰12刘亚兰12王雪萍12文杰12熊俊3冯永1,2,3
1中南大学湘雅医院耳鼻咽喉头颈外科(长沙410008)
2耳鼻咽喉重大疾病研究湖南省重点实验室(长沙410008)
3中南大学医学遗传学国家重点实验室(长沙410078)
4新疆医科大学第一附属医院耳鼻咽喉科(乌鲁木齐830011)
目的 分析一个常染色体显性遗传性耳聋家系临床及遗传学表型,并筛查常见耳聋致病基因。方法通过调查问卷、体格检查、听力学检测,完成该湖南籍耳聋家系的临床资料采集,绘制家系遗传图谱,分析其听力学及遗传学特征,对最常见的GJB2,SLC26A4和12S rRNA共3个耳聋基因八个位点以及线粒体DNA全组序列进行初步筛查。结果该家系共5代,现存家系成员35人,耳聋患者10人,除两人发病较晚,余均为自幼发病,听力曲线呈盆覆型,造成部分言语功能障碍,进展性加重,起初为中频受累,随着年龄的增长,以后逐渐累积高低频,表现为全频听力损失,发展为重度-极重度耳聋。对候选致病基因突变筛查,未发现致病突变。结论该耳聋家系符合常染色体显性遗传规律,进一步将通过新一代测序全外显子测序技术对其致病基因进行探索。
家系;常染色体显性遗传;遗传性耳聋
耳聋是人类最常见的感觉缺陷性疾病,每500个新生儿就有1个耳聋患儿[1],全世界范围内有2.78亿人构成的耳聋群体(http://www.who.int)。先天性遗传性耳聋主要是非综合征型耳聋(NSHL),约占70%,余约30%则为综合征型耳聋(SHL)。在发达国家,近80%的非综合征型耳聋是由遗传因素引起的,其中常染色体隐性遗传方式占60%-75%,余下患者中20%-30%为常染色体显性遗传,约2%为X连锁遗传和线粒体遗传[2]。常染色体显性遗传性耳聋(ADNSHL)一般变现为渐进性的语后聋,可以为以综合征型遗传病表型的一部分,例如Waardenburg综合征、Stickler综合征、Treacher Collins综合征,也可以是相对其他类型遗传耳聋不多见的非综合征型遗传耳聋,约18%遗传性耳聋表现为该种形式[3]。
随着基因测序技术的进步和发展,新一代的DNA目标区域捕获技术和高通量测序技术为检测耳聋突变基因提供了一种非常高效的手段,并极大地推动了耳聋基因的定位克隆工作的发展步伐,近5年来,应用新一代测序技术成功克隆了38个非综合征型遗传性耳聋致病基因(http://hereditaryhearingloss.org),尤其是2014年至2015年间,由于对测序技术的成熟掌握,后期数据分析的更为合理,耳聋基因的克隆迎来了突破性进展,两年间共发现了13个新的非综合征型耳聋基因——ADCY1、DCDC2、TBC1D24、NARS2、MET、TMEM132E、GRXCR2、EPS8、CLIC5、FAM65B、OSBPL2、HOMER2、COL4A6。因此我们完全有理由相信,随着新一代高通量测序技术成本的降低,应用范围的推广,测序技术及数据分析方法的进步,越来越多的耳聋基因将会被发现,并为进一步在临床耳聋基因诊断方面打下坚实的基础。
本研究报道一个湖南省的耳聋大家系,针对其临床表型及家系遗传特征进行分析,对常见耳聋基因及位点进行初步筛查,并制定后续运用新的遗传信息学技术对该耳聋家系致病基因研究的策略。
1 资料和方法
1.1家系资料的采集
家系资料的调查研究工作获得湘雅医院伦理委员会认可,由湘雅医院耳鼻咽喉头颈外科家系采集小组完成。本研究中发现的家系位于湖南省某市,先证者为1例17岁双耳重度感应神经性耳聋的女性患者,家系采集小组对该先证者及其各亲属成员进行了家系调查,所有家系成员均签署知情同意书,对家系中的16名成员完成问卷式调查资料、简易电耳镜检查、听力学检查、粗略智力评估、体格检查(包括专科体格检查和全身常规体格检查)等检查,采取外周静脉血3~8ml,提取基因组DNA并保存。
1.2遗传方式判断标准
目前国内外研究者对于耳聋有多种分型方法,根据听力损失的类型,可分为传导性、感音神经性以及混合性耳聋;根据发病年龄不同,可分为语前聋和语后聋;根据是否伴有其他组织器官的发育异常或功能障碍,可分为综合征型耳聋(SHL)和非综合征型耳聋(NSHL)。因此,结合家系遗传图谱及临床调查资料,分析其特异的遗传学规律及临床特点,从而判断家系的遗传方式。
1.3听力学检测及标准
本部分工作均由听力学专科技师以及临床专科医师依照规范标准完成。运用丹麦Madsen502便携式听力计对参与本研究的家系成员进行常规听力学检测,包括纯音测听、声导抗,先证者在湘雅医院耳鼻咽喉头颈外科学进行了听性脑干诱发电位(ABR)检测,根据WHO预防聋和听力损失项目报告(1997年,日内瓦),按较好耳0.5Hz~4KHz四个频率计算平均听阈,听力损失程度分级[4]:轻度(20~40dBHL);中度(41~70dBHL);重度(71~95dBHL);极重度(>95dBHL)。根据听力损失的频率特征分为:低频下降型(<0.5kHz);覆盆(中间)型(>0.5kHz<2kHz);高频下降型(>2kHz<8KHz)。这些标准有助于分析家系表型,进而指导遗传检测工作。
1.4候选耳聋基因筛查
目前耳聋基因的已定位了100多个NSHL基因座位(http://hereditaryhearingloss.org),对于耳聋基因定位克隆研究工作,其研究方法应灵活、特异且经济有效,我科临床定制遗传性耳聋基因检测试剂盒(英盛生物耳聋基因检测试剂盒)可通过荧光PCR技术特异性对中国人群最常见的三个遗传性耳聋致病基因(GJB2、SLC26A4、12SrRNA)8个最常见耳聋基因突变位点进行定性筛查,运用该试剂盒对先证者进行致病突变筛查,并利用Sanger测序对线粒体基因全序列进行筛查,排除常见已知致病突变位点。
2 结果
2.1家系基本资料
该家系5代居住于湖南省某市,现存家系成员35人(17男,18女),其中确诊患者10人(5男5女),耳聋患者年龄最大者90岁,最小者17岁,均以耳部症状为单一症状,无噪声接触史及耳毒性药物用药史,均无前庭功能障碍,未见明显其他器官、系统异常,其中8人自幼出现听力损失,余2人发病较晚。先证者为Ⅳ-15,女,17岁,因双耳听力下降配戴助听器效果不佳入院就诊,纯音测听示双耳对称性、重度感音神经性耳聋,无眩晕史。双耳听力曲线均为呈覆盆型;双耳声导抗A型曲线;ABR示:双耳80dBHL诱出反应波。家系内成员均生活在同一环境,排除非遗传性因素对耳聋患者听力损失的影响。家系患者资料详见下表2.1
2.2家系听力学特征及遗传性特点
2.2.1家系听力学特征
该家系第Ⅱ代有血缘关系成员共有4人,明确耳聋患者2人,均为自幼耳聋,可言语,听力渐进性下降,极重度感音神经性耳聋,听力曲线为岛状型(残余听力)。第Ⅲ代家系有血缘关系的成员6人中,耳聋表型确认者3人,均为自幼耳聋,可言语,Ⅲ-4、Ⅲ-9为重度感音神经性聋,全频受累,Ⅲ-7为重度感音神经性聋,交流困难,各频段均受累,听力曲线呈平坦型,第Ⅳ代有血缘关系的成员12人,明确耳聋患者4人,三代现存家系成员的男女性患者比例为4:5,均为自幼发病,但均存在不同程度言语发育障碍,家系第Ⅴ代家系成员中,Ⅴ-2、Ⅴ-5虽然父亲均为耳聋患者,各自只生育一个女儿,而该两名儿童均无耳聋,言语发育正常,余两名耳聋患者尚无子女,故依据现有资料不足以判断家系是否存在发病逐代提前或延后的趋势,家系部分患者纯音听阈检测结果见图2-2。家系中有两名成员表现特殊:
Ⅲ-11,耳聋患者Ⅱ-4的子代,听力检测发现双侧对称性轻度听力损失,发病年龄较晚,48岁出现渐进性听力下降,以4KHz及8KHz高频损失为主,听力曲线表现为下降型,全身及专科体格检查未发现异常,否定耳毒性药物及噪声接触史,其言语发育正常,且两个子女均无耳聋表现,而家系内耳聋成员绝大多数为自幼发病,综合其病史和听力损失特征,由此推测Ⅲ-11耳聋表型可能并非该家系致聋基因引起,不排除年龄相关性感音神经性听力损失或其他耳聋基因导致的听力损失。
Ⅳ:7,其母亲为耳聋患者Ⅲ-4,从17岁开始出现听力下降,渐进性加重,听力检测为双侧对称性、中度感音神经性聋,听力曲线呈特异性覆盆型,通过病史资料采集及体格检查,否定了耳毒性药物用药史及噪声接触史,无其他组织器官发育及功能异常,明确为非综合征型感音神经性耳聋患者,同其弟弟Ⅳ:7耳聋表型相似,言语发育无明显障碍,考虑遗传性耳聋异质性,故成员Ⅳ:7耳聋症状仍可能是本研究家系致聋基因引起。
2.2.2家系的遗传学特征
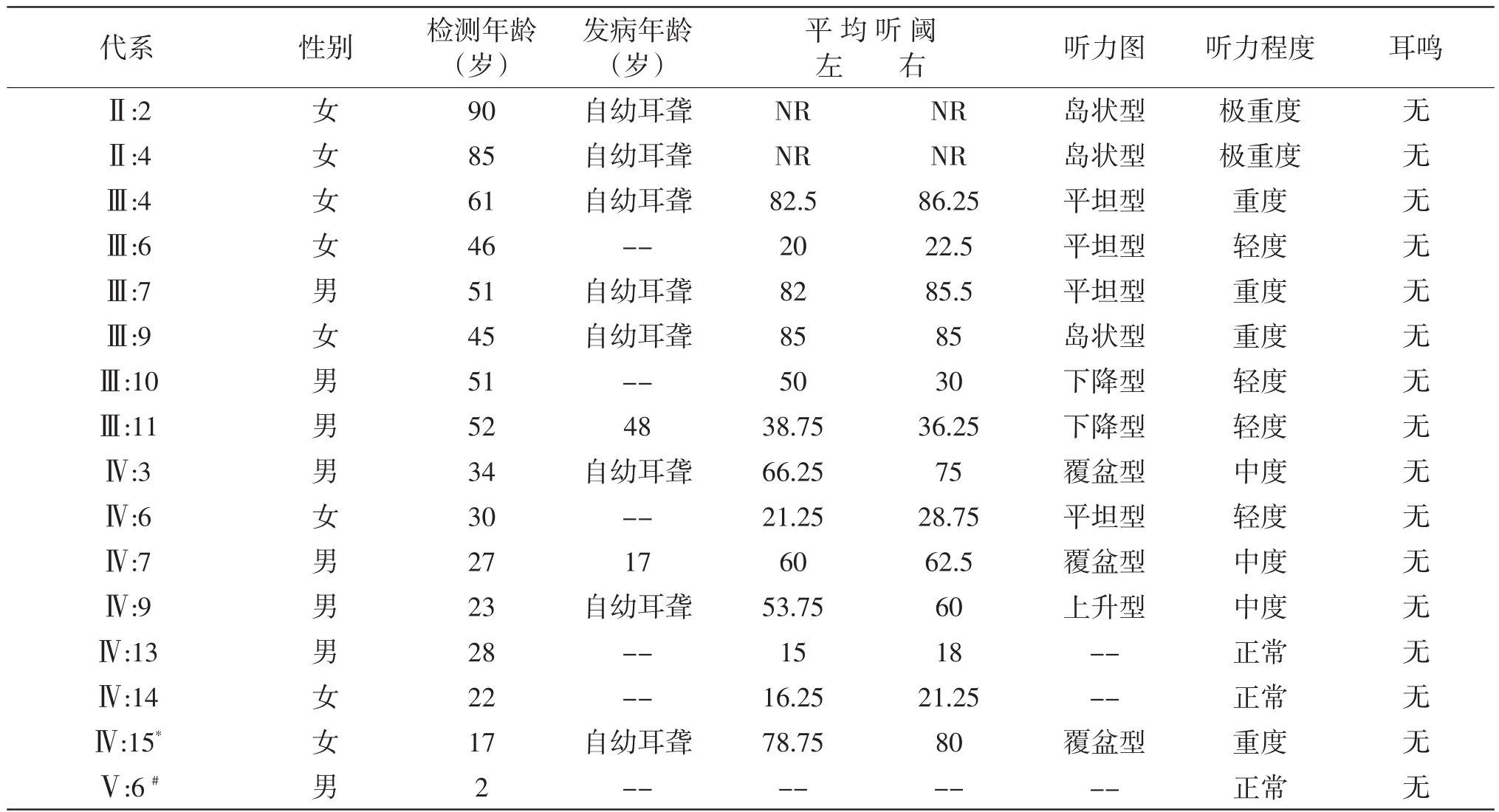
表2.1 家系患者临床表现
根据本家系研究采集小组收集到的信息资料和调查结果,应用cyrillic3.0软件绘制系谱图(见图2-3),通过系谱图可知,家系共5代,3代连续发病,现存家系成员35人,主要为第二至第四代成员,由于Ⅰ代家系成员去世已久,现存家系成员对其发病情况不清楚亦无法追溯,第Ⅱ代家系直系血缘关系成员,两名女性(Ⅱ-2、Ⅱ-4)均发病,患者Ⅱ-2其后两代子女均出现了耳聋患者,男女比例4:3。综上所述,该家系连续三代中均有发病患者,男性或女性均可患病,且男女患者比例相近,以上特征符合典型的常染色体显性遗传模式。
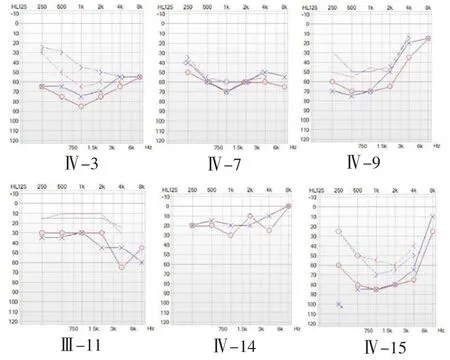
图2.2 家系部分成员纯音听阈图
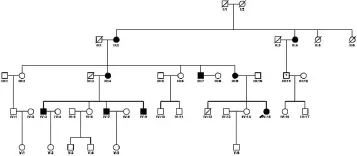
图2.3 耳聋家系系谱图
2.3候选基因突变筛查结果
家系中先证者耳聋基因检测试剂盒筛查结果均为阴性,其位点包括:GJB2:c.35delG、c.176-191del16bp、c.235del C、c.299-300del AT;SLC26A4:IVS7-2A>G、c.2168A>G;12SrRNA:m.1555A>G、m.1494C>T共3个基因8个最常见耳聋基因突变位点。线粒体全基因序列Sanger测序发现31个多态位点。
3 讨论
遗传性听力耳聋中基因型跟表型之间关系密切,从听力损失角度来看,某种听力损失表型往往一定程度上反映相关基因突变的基因型,故而越了解这种相互关系,我们就越容易推测某特定听力损失类型的基因型。非综合征型常染色体显性遗传性耳聋(DFNA)中,绝大多数表现为差异不明显的高频听力下降,而低频和中频听力损失表型相对少见。在家系收集过程中尽可能详尽的、仔细的记录好所有成员临床病史资料,对家系的表型信息和遗传方式的分析,以及进一步候选基因筛查和定位研究显得尤为重要。
本研究家系根据遗传图谱分析符合孟德尔常染色显性遗传,根据病史及听力检测可得出,该家系患者成员语言部分发育受阻,但均无智力异常,表明发病年龄为学龄期或学语前,虽有逐渐加重趋势但是进展缓慢,听力曲线呈覆盆型,均为自幼发病,造成部分言语功能障碍,起初为中频受累,随着年龄的增长,以后逐渐累积高低频,表现为全频听力损失,发展为重度-极重度耳聋。
候选基因筛查是目前广泛应用的基因克隆研究策略之一,假设耳聋家系遗传模式的基础上,根据耳聋基因的表型特性以及耳聋基因的流行病资料,选取最常见、致聋率最高的耳聋基因,作为待排除的家系致聋候选基因,技术上方法包括Sanger测序、微卫星标记、高通量捕获、荧光探针等,本研究家系中我们对先证者运用我科临床耳聋筛查试剂盒——英盛生物耳聋基因检测试剂盒,对中国耳聋人群最常见、检出率最高的GJB2、SLC26A4、12SrRNA共三个基因的8个位点进行筛查排除,根据家系特征,男性患者后代无耳聋病人,仍有母系遗传可能性,故我们运用Sanger测序对线粒体全组序列完成筛查,未发现报道耳聋突变,初步排除线粒体遗传可能,家系中第Ⅳ-3、Ⅳ-7只育有一女,概率可能显性耳聋基因未能表现出来,并且Ш-7、Ⅳ-15及Ⅳ-9尚无子嗣,故目前尚不足证据说明男性患者不会把耳聋表型先下传递给后代,以及该家系发病年龄是否延后趋势,结合临床资料及遗传图谱,本研究家系为常染色体显性遗传非综合征型耳聋家系。
截至目前,已报到常染色体显形遗传非综合征型耳聋基因中以中频下降为主的有DFNA10 (EYA4)、DFNA13(COL11A2)、DFNA8/12(TECTA)共3个基因。EYA4基因是在一个四代美国耳聋家系,通过连锁分析最终定位在6p22-23染色体上的DFNA10位点[5],cDNA全长1920bp,共21个外显子,可编码639个氨基酸。大鼠模型显示Eya4主要表达在耳蜗血管纹和前庭膜。EYA4基因突变引起的耳聋主要表现为30左右开始高频和中频听力损失,逐渐累积全频,听力曲线表现为下降型或平坦型[6]。
COL11A2基因位于6p21.3,全长28000bp,包含有66个外显子,DFNA13位点上COL11A2基因的突变导致听力曲线呈特征性的U型,在老年型耳聋发病年龄之前,患者的中频听力下降进展缓慢[7]。该型大部分的耳聋患者在20~40岁开始发病,部分耳聋家系中实际听力损失可追溯到学语前。该耳聋基因的小鼠模型发现小鼠耳蜗的Corti器盖膜变薄、结构疏松,并且研究发现其归因于Ⅱ型胶原纤维并非像正常的平行均匀的方式排列,而是呈一种无序的杂乱方式排列[8]。Col11a2基因突变可能是通过这种机制影响了盖膜的正常功能。
TECTA基因是通过一个4代先天性、常染色体显性、非进行性奥地利耳聋家系定位在染色体11q22.24上36cM区域[9],TECTA基因的透明带区域的突变可以引起学语前、非进行性、中频为主的听力损害,TECTA基因的纯和突变还可以导致常染色体隐性、非综合征型重度至极重度的听力损失(DFNB21)[10],TECTA(DFNA8/12)编码α-tectorin蛋白,该基因错义突变可导致α-tectorin蛋白透明带区域的高度保守型氨基酸的异常改变,突变蛋白可通过显性负性效应影响盖膜正常结构,影响了毛细胞纤维束对声振动的机械传输的效率[9]。
以上三个基因虽为首选候筛基因,但每个基因都很大,为了节约成本,缩短周期,随着国际人类基因组计划(HGP)、国际千人基因组计划、人类基因组单倍型图计划、基因组关联计划等项目相继开展和完成,以及生物信息学的高速发展,第二代测序技术衍生的全基因外显子组测序(WES)技术得到了广泛应用,目前已成为遗传性耳聋致病基因筛查和鉴定工作的主要技术。WES技术具有高效、快速的特点,本课题组拟选取表型明确且遗传间隔距离较大的Ⅳ-15和Ⅳ-3两名患者进行全基因外显子组高通量测序。以期找到该家系耳聋致病基因。
1MORTON CC,NANCE WE.Newborn Hearing Screening-a Silent Revolution.N Engl J Med,2006:354(20):2151-2164.
2MORTON NE.Genetic Epidemiology of Hearing Impairment.Ann N Y Acad Sci,1991:630(1):16-31.
3SHEARER AE,SMITH RJ.Genetics:Advances in Genetic Testing for Deafness.Curr Opin Pediatr,2012:24(6):679-686.
4王秋菊.《关于非综合征型遗传性听损伤家系遗传学及听力学描述术语建议案》.中华耳科学杂志.2003,1(04):46-51.
5VERHOEVEN K,FAGERHEIM T,PRASAD S,et al.Refined Localization and Two Additional Linked Families for the DFNA10 Locus for Nonsyndromic Hearing Impairment.Hum Genet,2000:107(1):7-11.
6WAYNE S,ROBERTSON NG,DECLAU F,et al.Mutations in the Transcriptional Activator Eya4 Cause Late-Onset Deafness at the DFNA10 Locus.Hum Mol Genet,2001:10(3):195-200.
7DE Leenheer EM,KUNST HH,MCGUIRT WT,et al.Autosomal Dominant Inherited Hearing Impairment Caused by a Missense Mutation in Col11a2(DFNA13).Arch Otolaryngol Head Neck Surg,2001:127(1):13-17.
8MCGUIRT WT,PRASAD SD,GRIFFITH AJ,et al.Mutations in Col11a2 Cause Non-Syndromic Hearing Loss(DFNA13).Nat Genet,1999:23(4):413-419.
9KIRSCHHOFER K,KENYON JB,HOOVER DM,et al.Autosomal-Dominant,Prelingual,Nonprogressive Sensorineural Hearing Loss:Localization of the Gene(DFNA8)to Chromosome 11q by Linkage in an Austrian Family.Cytogenet Cell Genet,1998:82(1-2):126-130.
10MUSTAPHA M,WEIL D,CHARDENOUX S,et al.An Alpha-Tectorin Gene Defect Causes a Newly Identified Autosomal Recessive Form of Sensorineural Pre-Lingual Non-Syndromic Deafness,DFNB21.Hum Mol Genet,1999:8(3):409-412.
接673页版社,1998:764-765
3Margolis RH,Bass-Ringdahl S,Hanks WD,et al.Tympanometry in newborn infants:1KHz norms.The American Academy of Audiology.2003,14:383-392.
4Jerger J.Clinical experience whith impedance audiometry.Arch Otolaryng.1970,92:311-24.
5陈文霞,徐正敏.125例新生儿的鼓室导抗测试结果分析.听力学及语言疾病杂志.2009,17(6):546-548.
6Rosenfeld RM,Culpepperl L,Doyle KJ,et al.Clinical practice guideline:otitis media with effusion.Otolaryngol Head Neck Surg.2004,130(5):95-118.
7Purdy SC,Williams MJ.High Frequency Tympanometric:a valid and reliable immittance test protocol for young infants?New z Audiologic Soc Bull.2000,10:9-24.
8Chang C,Pedler K.Ear examination-A practical guide.Aust Fam Physician.2005,34:857-862.
9Hunter LL,Margolis RH.Multifrequency tympanometry:current clinical application.[J]the American Academy of Audiology.1992,1:33.
10Baldwin M.Choice of probe tone and classification of trace patterns in tympanometry undertaken in early infancy.Int J Audiol.2006,45:417.
11黄丽辉,马潇然,王硕等.检测婴幼儿中耳炎的听力学方法敏感性比较.听力学及言语疾病杂志.2010,18:553.
12Cone-Wesson B,Ramirez GM.Hearing sensitivity in newborns estimated from ABRs to bone conducted sounds.Am Audiol.1997,8:299-307.
13韩德民,许时昂.听力学基础与临床,北京:科学技术文献出版社.2003:297-297.
14Spector GJ,Ge XX.Development of the hypotympanum in the human fetus and neonate.Ann Otol Rhinol Laryngol Suppl.1981,90:6-2.
15倪道凤.婴幼儿中耳炎的诊断与治疗.临床耳鼻咽喉科杂志.2005,19:577.
16罗仁忠,温瑞金,王美芬等.6岁以内小儿鼓室压与静态声顺值的特点.临床耳鼻咽喉科杂志.2002,16(10):524-525.
(收稿日期:2015-11-2审核人:于黎明)
Analysis of Clinical and Genetic Characteristics of a Chinese Pedigree withAutosomal Dominant Hereditary Nonsyndromic Hearing Loss
Niu Zhijie1,2,Sun Jie1,2,4,Mei Lingyun1,2,Jiang Lu1,2,Cheng Hongsheng1,2,He Chufeng1,2,Liu Yalan1,2,
Wang Xueping1,2,2Wen Jie1,2Xiong Jun3,Feng Yong1,2,3
1 Department of Otolaryngology-Head and Neck Surgery,Xiangya Hospital,Central South University,Changsha,410008,China
2 Key Laboratory of Major Otolaryngology Diseases Studies of Hunan Province,Changsha,410008,China
3 State Key Laboratory of Medical Genetics,Central South University,Changsha,410078,China
4 Department of Otorhinolaryngology,First Affiliated Hospital of Xinjiang Medical University,Urumqi,830011,China Corresponding author:Feng Yong Email:fengyong_hn@hotmail.com;
Objective To study clinical and genetic characteristics of a large Chinese pedigree with autosomal dominant nonsydromic hearing loss,and to report screening results in this pedigree on common causative genes.Methods After obtaining informed consents from all participants,deafness-questionnaires were administered to collect detailed medical histories.Clinical presentations,otoscopy findings and pure-tone audiometry were used to rule out syndromic hearing loss.The pedigree was plotted based on the genetic and audiology characteristics of this family.A deafness-screening kit was used to screen for eight common mutations of three deafness genes(GJB2,SLC26A4 and 12S rRNA)and the whole MTDNA to exclude known pathogenic mutations.Results Thirty five family members were alive in this five-generations family with autosomal-dominant hearing loss,and 10 were found to be hearing-impaired.Most of the patients showed moderate to severe pre-lingual sensorineural hearing loss affecting predominantly mid frequencies with slow progression to all frequencies.Audiometric configurations were characterized by“U”shaped or“island”patterns.No specific causative mutations were identified by screening.Conclusions Pedigree analysis in this family confirms an autosomal dominant inheritance pattern,in which affected members show pre-lingual,symmetry,gradually-progressive hearing impairment.Future studies using whole-exome sequencing is planned to further explore disease-causing genes in this family.
Pedigree;Autosomal dominant inheritance;Hereditary deafness
R764.43
A
1672-2922(2015)04-662-5
2015-10-8审核人:郭维维)
10.3969/j.issn.1672-2922.2015.04.021
国家重大科学研究计划项目(Grant No.2014CB541702,2014CB943003)、国家自然科学基金项目(Grant No.81170923,81470705,81300833)及湖南省自然科学基金(Grant No.14JJ7009)
牛志杰,在读博士研究生,研究方向:耳科学
冯永,Email:fengyong_hn@hotmail.com
