基于染色质交互数据的基因组组装方法
2015-09-14陶婧芬谢婷郑觉非杨庆勇张红雨
陶婧芬 谢婷 郑觉非 杨庆勇 张红雨
DNA是生物体遗传信息的主要载体,高质量的基因组参考序列是现代遗传学、分子生物学等现代生物学科的重要基础。因此,基因组测序对探索与认识生命本质等基础生物科学研究、人类重要遗传病防治及动植物遗传育种等应用性研究均具有十分重要的意义。
1 Scaffolds锚位逐渐成为制约高质量全基因组序列获得的主要挑战
基于二代测序技术,又称下一代测序技术(Next generation sequencing,NGS)的全基因组测序工程一般包含两个部分:拼接和组装,前者是将二代测序技术产生的DNA测序片段(Reads)拼接成小的重叠群(Contigs)的过程,后者是将拼接阶段产生的重叠群组装成长序列片段(Scaffolds),以及将长序列片段定位到染色体上的过程。伴随着DNA测序技术的不断推陈出新[1]和单位测序成本的大幅度降低[2],如何准确、高效、快速地将scaffolds定位到染色体上逐渐成为高质量全基因组序列获得的主要挑战。
1.1 基因组测序现状
得益于DNA测序技术飞速发展,不断有新的物种基因组被测序,继而由测序的片段组装出相对完整的基因组序列。现有的基因组测序工程主要借助于全基因组鸟枪法(Whole genome shotgun,WGS)的策略[3],其原理是将基因组打断成小片段,随后将片段克隆到载体上组建重组克隆群并测序以获得用于组装的序列。这种方法克服了大片段克隆分别测序(Clone-by-clone,CBC)策略难以分离并克隆着丝粒等区域的缺陷。随着高通量测序技术的发展,WGS策略以更低的成本以及更高的效率成为近年来大多数测序工程的首选。截止到现在,GenBank中采用WGS方法进行测序组装的项目已有42 925个(http://www.ncbi.nlm.nih.gov/assembly/,统计日期:2015年7月20日)。
尽管在测序和拼接技术日趋完善的今天,绝大多数物种的组装结果仍然不够完整且存在不少组装错误[4],并且很多已被测序物种的参考序列信息仍以零散的序列片段的形式存在。NCBI中的数据统计显示(http://www.ncbi.nlm.nih.gov/assembly/,统计日期:2015年7月27日),仅有26.6%的植物、12.0%的动物和15.4%的真菌基因组完成了染色体水平的组装(表1)。由此可见,基因组组装大多仅仅停留在长序列片段(BAC和/或scaffolds,下文统称为scaffolds)的水平,而确定scaffolds在染色体上的具体位置逐渐成为染色体水平的参考序列获得的限制环节。
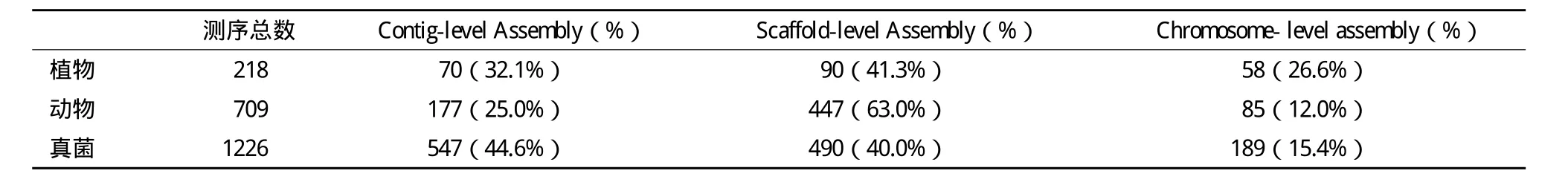
表1 植物、动物、真菌中基因组组装情况
1.2 传统scaffolds锚位方法存在的主要困难
传统的scaffolds锚位方法主要分为两个大类,基于物理图谱的方法和基于遗传图谱的方法。前者是通过序列或序列特征的重叠关系来确定DNA片段的位置,后者是利用减数分裂时期的姊妹染色单体联会后不同DNA片段共交换的频率来判断DNA片段的相对位置。由于这两类方法都包含大规模文库或群体构建、筛选等一系列复杂的实验过程,其所需成本、结果的精度、准确性等在很大程度取决于实验的设计和实施,在实际的scaffolds锚位的过程中主要存在以下难点。
1.2.1 大片段文库构建难度大 构建大片段的BAC文库是基于物理图谱锚位scaffolds方法的限制性环节。而传统的基于遗传图谱锚位scaffolds的方法对片段长度(如scaffolds N50)要求较高,为了提高scaffolds的长度,一般需要构建大片段mate pair测序文库。大片段文库构建的整个操作流程相比普通实验更复杂且对实验经验的要求更高[5]。在文库构建过程中将基因组片段插入载体中,不同物种基因组的重复度高低等指标会影响大片段文库插入片段长度的目标值;而插入片段越长、连接率越低、构建成功率则越低;再者,随着插入片段的增大,文库冗余率的升高等均会影响大片段文库最终有效数据的产出。因此,对于大部分分子生物学实验室,都不具备构建高质量、低冗余率大片段文库的技术条件,目前该系列实验仍存在诸多困难。
1.2.2 成本较高 一方面,传统的scaffolds锚位方法通常需要构建遗传群体或者基因组文库。对于植物来说,其生长周期一般都超过3个月甚至更长,并且经常受制于种植季节,构建作图群体一般就需要1-2年,整个过程将会消耗更多的时间。另一方面,传统的组装方法在前期进行大规模的实验,这需要消耗大量的人力成本和物力成本来建立遗传分离群体和标记基因型分析。特别是为了提高定位精度,需要进一步提高有效标记密度时(即获得更多的交换单株),随着作图遗传群体的扩大,需要消耗大量的人力物力[6]。
1.2.3 误差偏高 传统的scaffolds锚位方法一般涉及大规模田间种植和分子生物学实验,在复杂繁琐的实验过程中,多个环节实验不可避免地导致实验误差和随机偏差的积累,同时也更容易引入人为误差和系统误差。
2 高通量染色质构象捕获技术为scaffolds快速锚位提供了契机
2.1 染色质构象捕获技术
染色质构象捕获(Chromosome conformation capture,3C)技术原本用于研究基因表达时染色质的空间构象[7]。该技术利用了连接反应倾向于发生在物理上相互靠近的DNA片段之间(即邻近连接原则),然后利用PCR对模板数量的敏感性,迅速准确地抓出与目标区域相互靠近的DNA片段。
Hi-C(High-throughput chromosome conformation capture)技术是由3C[6]技术发展而来,结合了生物素标记筛选和二代测序技术,通过交联、酶切、连接等步骤,实现全基因组范围内染色质交互的高通量检测。2009年,Job Dekker的研究小组在3C技术的基础上开发出全基因组范围的染色质构象捕获技术(Hi-C),获得了分辨率为1 Mb的交互图谱并模建出核内染色质的三维立体模型[8]。研究人员通过化学手段固定住蛋白与核酸或蛋白与蛋白之间的接触。随后将DNA片段化,并将相互联系的DNA连接在一起。最终对所有区域间的接触次数进行统计,绘制出交互矩阵,便可估算出三维状态下任意两个区域相隔距离。
目前,染色质交互数据在酵母、人类、小鼠、果蝇和拟南芥等物种中均有过报道。其中人类的染色质交互数据达到了1 kb的分辨率[9],精细程度深入到了单基因水平。
2.2 染色质构象捕获用于基因组组装研究的现状
Hi-C技术传统应用于研究与特定蛋白质因子作用的染色质组和全基因组范围内染色质组的互作[10]。同时,Hi-C产生了大量的染色质交互数据,根据这些染色质交互数据,可以重建染色质的三维结构[11]。真核生物的基因组在细胞核中以染色质的形式存在,基因组的复制、转录、调控、DNA突变、长链非编码RNA的传播和胚胎发育等生物功能与其三维结构密切相关[12]。三维结构的重建,为我们更加系统地了解染色质的调控功能提供结构依据[13]。
此外,Hi-C技术所揭示的染色质片段间的交互强度呈现出随距离衰减的规律[8]。正是这一规律,催生出了“基于Hi-C技术组装基因组”这一新的研究领域。与传统的遗传定律相类似,这一规律可以用来判断scaffolds的分群及相邻关系。具体而言,“染色体内交互高于染色体间交互”可以指导核酸片段的染色质分群,“同一染色体上近程交互高于远程交互”则可以引导核酸片段的排序和定向。
目前Hi-C技术应用于基因组组装的物种主要包括人类、小鼠、果蝇、拟南芥、酵母以及其他微生物和微生物群落。2013年,Job Dekker等人[14]通过整合Hi-C数据、鸟枪法测序序列以及短序配对(Short jump mate-pair)文库序列定位了人类基因组中65个尚未锚定到染色体上的重叠群,与其他方法得出的结果有83.78%相吻合。其中挂载到染色体的准确率为99.80%。Burton等[15]将这种方法应用到了人、小鼠和果蝇的全基因组de novo组装当中,占人类和小鼠序列总长超过98%的scaffolds被用于分组、排序和定向,正确率达到90%以上。在果蝇中,虽然原始鸟枪法得到的scaffolds 质量与人类和小鼠相比较差,分组和排序的scaffolds利用率能达到81.2%和82.0%,scaffolds定向的正确率高达93.9%。Marie-Nelly等[16]用这种方法填补酿酒酵母基因组组装中的缺口(gap),随后又用它来组装里氏木霉菌基因组。Burton小组[17]将Hi-C技术与宏基因组学相结合,在微生物群落的物种鉴别以及单个物种基因组组装上都取得了很好的效果。而Putnam等[3]利用体外模拟体内DNA互作获取的染色质信息组装美国短吻鳄基因组,其中,在人类中68.9%测序读长的比对质量超过了20;在美国短吻鳄中1298个测序读长覆盖度达到90%,一致性达到95%,都取得了较好的应用(表2)。
本课题组利用此方法来组装拟南芥基因组。利用有效的Hi-C交互数据,将总长度为112.61 Mb的 1705个 scaffolds[18]进行分群,其中 1350个scaffolds(占总长的97.12%)能够被准确地分配到其相应的染色体上。基于染色体局部交互信息,对551个的scaffolds进行了排序和方向确定,其中516个(占总长的92.29%)scaffolds能够被准确排序和确定方向[19]。

表2 Hi-C数据从头组装基因组结果汇总
3 基于体内染色质交互数据组装基因组的主要流程
3.1 实验操作
现有的获取染色质体内交互数据的技术有很多,都是基于染色质构象捕获技术(3C)发展而来,而应用于组装最多的是Hi-C技术。Hi-C实验主要的原理是甲醛能在常温下与氨基或羟基发生化学反应,将蛋白与DNA或蛋白与蛋白之间的物理接触“固定”下来。Hi-C技术的大致流程为:通过甲醛交联固定,将细胞内由蛋白质介导的空间上邻近的染色质片段进行共价连接。甲醛交联后加入特定的限制性内切酶进行酶切。酶切后的黏性末端利用核苷酸补平,用于补平的其中一种核苷酸(如C)用生物素标记。之后在非常稀释的环境中,加入连接酶连接平末端形成分子内连接,原有的酶切位点丢失,取而代之的是新的酶切位点。最后将连接的DNA进行纯化后超声破碎,并用生物素亲和层析将生物素化的DNA片段分离出来,加上接头通过高通量双末端测序检测交互的 DNA片段[8]。
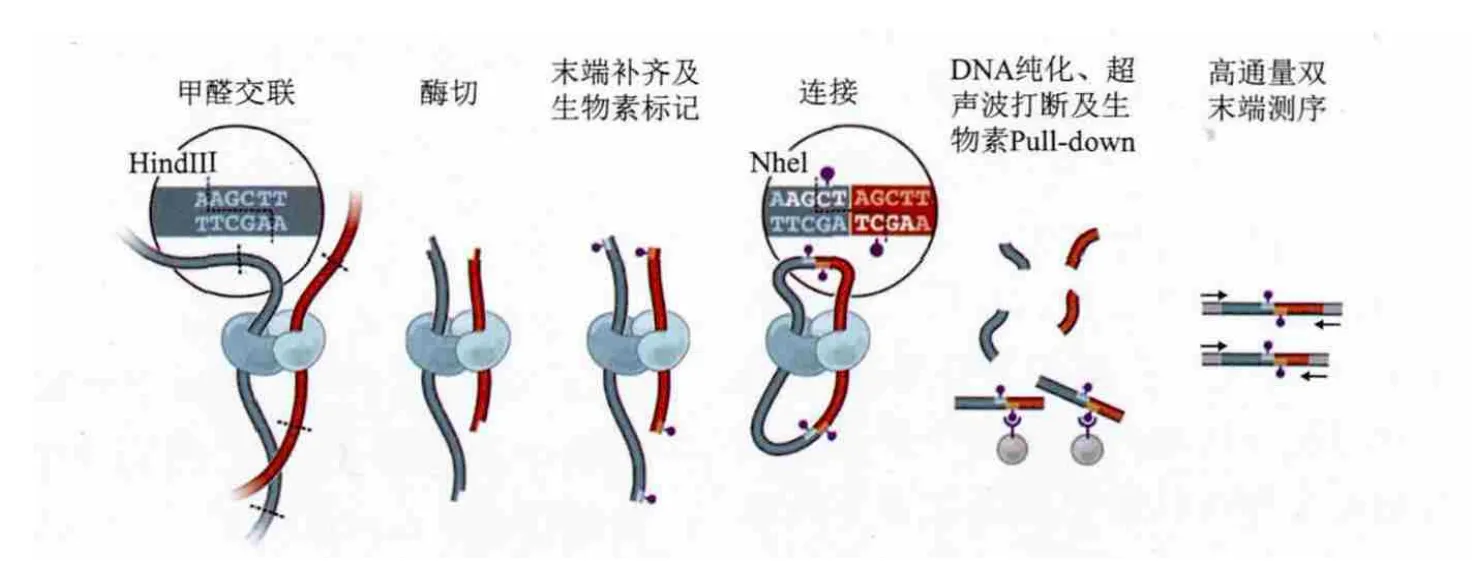
图1 Hi-C测定染色质交互的基本原理及技术流程[8]
3.2 数据分析
3.2.1 数据的比对、去噪和校正 Hi-C实验得到的原始染色体交互数据中具有大量的噪声,因此,在基因组组装前必须对原始数据进行处理。通过测序平台获得的原始交互数据是双端测序数据,即pairend reads。与其他二代测序实验一样,必须先检测测序的质量。因为实验操作中可能因为条件控制而导致实验差错,对于建库测序的结果,需要用相关的测序数据质量控制软件(如FastQC)衡量数据的可利用性。
在确定获取的数据质量之后,需要将双端测序结果比对到参考基因组上。可直接使用短序列比对软件设置相关参数进行比对,也可以运用迭代增加mapping reads长度的比对算法[20],以便最大限度增加数据的利用率。
最后,Hi-C实验的各个操作步骤会引入各种各样的噪声,包括PCR重复、随机打断、自连接、随机连接等[20-22],所以必须根据数据特征对这些噪声进行过滤。同时,序列本身的特征如GC含量、酶切位点频率[20,21]等都会对交互数据产生影响,因此通常还要对得到的原始交互数据进行迭代校正(Iterative correction and eigenvector decomposition,ICE)[20]。通过上述质量控制步骤后,我们可获得用于基因组组装的Hi-C交互数据。
3.2.2 构建交互矩阵和挂载scaffolds 利用去噪校正之后的交互数据,构建染色质交互矩阵。如果有两个以上技术重复,还需要检验交互矩阵的皮尔森相关性。
针对其染色质三维空间结构特征,选取合适的聚类模型将未定位scaffolds锚定到染色体上,并采用相应的排序算法确定挂载scaffolds的正确顺序和方向,组装出染色体水平的全基因组序列(图2)。目前基于染色质交互数据进行基因组组装的几个软件都是按染色质三维空间交互规律开发的(表3),每个软件在分组、排序和定向中采用的算法不同,使得不同软件的参数设置也有所区别。研究者在基因组组装过程中需要根据自身研究目标和数据特征来选择不同的组装软件。
4 染色质构象捕获用于基因组组装研究的主要优缺点
与传统的组装方法相比,基于染色质交互数据确定scaffolds在染色质上的具体位置具有以下三个方面的优势:
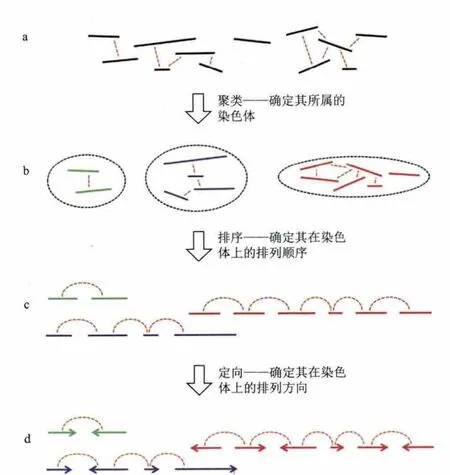
图2 利用染色质交互组装基因组示意图(以LACHESIS软件为例[15],有改动)

表3 基因组装的相关软件对比
(1)利用染色质交互的reads分布来判定scaffolds的相对位置,具有更高覆盖率和特异性。基于染色质交互的组装方法的reads长度是一般遗传标记的4-5倍,这使得其具有更高的位点特异性。此外,利用全局染色质捕获技术能获取所有的scaffolds片段的交互信息,因此绝大部分scaffolds都能被组装。
(2)基于单一株系染色质交互规律的组装方法,是利用scaffolds在体内染色质相互作用的分布特征来判定染色体片段之间的邻接关系,比利用亲本后代遗传连锁交互的组装方法更为直接和可靠。同时,它避免了繁琐的群体构建工作,在极大程度上减少了实验误差、系统偏差及机械混杂等不可控因素的干扰。
(3)基于染色质交互的组装方法要求的基础数据为进行基因组测序材料的单一株系Hi-C交互数据,整个过程无需构建庞大的遗传群体和进行大规模的基因型分型工作。相比之下,Hi-C技术实验周期短、实验规模小,节约了时间和成本。
由于Hi-C技术是以二代测序为基础的,在基于交互组装基因组的过程中,二代测序技术中存在的偏好和问题很有可能被引入到基因组组装过程中。首先,位于着丝粒和端粒附近的序列往往是高度重复的,二代测序从根本上是无法确定其具体的序列信息的,也就很难对其完成组装。也就是说基于交互组装基因组只能在原有的基础上提高基因组组装的正确率和完成率,而无法使其达到100%。其次,由于Hi-C技术本身分辨率的限制,使得组装无法更加精细,这一缺点有望在原位Hi-C中得到改进。再者,基于染色质交互组装基因组方法的主要理论基础是“近程交互高于远程交互”这个一般性、全局性的规律,而事实上在特定的小区域(如着丝粒、断离及拓扑相关结构域(Topologically associating domain,TAD)等,这一规律并不总是成立[23]。综上所述,在scaffolds片段较小(< 5 kb)、高度重复序列区域等因素都可能直接导致scaffolds锚位准确性和覆盖率降低。
5 基于染色质交互组装基因组的应用前景和展望
染色质构象捕获技术表明,高等生物细胞核内染色质片段间的交互不是随机、杂乱无章的,而是遵循着“染色体内交互高于染色体间交互,近程交互高于远程交互”这一基本规律的。从生物学意义上讲,这一规律反映了高等生物染色体三维结构形成的内在模式;在本文中我们展现了将这一规律应用于基因组组装的潜力。基于Hi-C技术进行基因组组装的方法具有实验操作简单、周期短、成本低的优点,能够在有限的人力物力条件下获得高覆盖率和准确率的参考基因组。即使与目前正在兴起的三代测序相比,该方法在成本上仍然具有相当的优势。
相比传统的基因组组装的方法,以染色质相互作用为基础的组装拥有较高的特异性和不依赖于遗传群体等特点,可能更适合复杂的基因组组装。同时,Hi-C实验简单并且有较短的时间周期和较低的成本,这使得基于染色质交互的组装方法有望获得更广泛的应用。因此,基于染色质交互组装的方法在实验设计、测序策略及算法等层面都存在较大的发展空间。
5.1 应用前景
基于染色质交互的组装方法从DNA片段交互频率与染色体内部结构之间的关系出发,避免了群体规模和交换频率这两个问题,可以与遗传图谱方法互相补充,并且极大地节省了时间和成本。与经典可靠的物理图谱组装方法相比,基于交互数据组装基因组在实验规模、时间消耗和人力物力等方面均远远小于物理图谱方法。结合该方法的优势,我们认为基于染色质交互数据的组装方法可在以下四个方面获得较大应用前景。
第一,测序基因组的进一步完善。目前最为常用的是使用遗传连锁图来挂载和确定contigs/scaffolds的染色体位置,但受限于物种群体规模和交换频率,仍然有许多contigs/scaffolds不能确定染色体位置,因此,继续使用遗传图谱方法来确定这部分序列将会花费巨大人力和物力。而利用基于染色质交互数据的方法,可用于挂载未挂载到染色体上的scaffolds的锚位和方向确定,从而提高已测序完成的基因组参考序列的完整性。
第二,高度杂合的植物基因组从头组装和完善。由于多年生物种的杂合度高,群体的构建具有很大的困难,这就限制了基于遗传连锁图谱挂载scaffolds的可行性、精度和准确性。而基于染色质交互组装的方法不依赖于遗传群体,仅需测序亲本的少量组织样品即可开展。因此,我们认为这种不依赖遗传群体的方法能应用于杂合度较高的植物基因组组装和完善中,并能获得更加真实和完整的参考序列。
第三,多倍体物种基因组的进一步完善。经典的基于遗传图谱挂载染色体的方法主要是通过SSR或SNP探针等遗传标记来反映同源染色体之间的遗传交换,然后利用标记之间的遗传连锁关系来判断染色体片段的相邻关系。而基于Hi-C的方法是利用reads之间交互的强弱来判断其染色体片段的相邻关系。相比前者,基于Hi-C的方法的reads长度是SSR、SNP等遗传标记的4-5倍,这使得其具有更高的位点特异性。因此,我们认为这种高特异性的方法应用在基因组相对复杂,多倍体现象十分普遍的物种、尤其是植物中具有更大的优势。
第四,具有重要科研、生态价值或区域特色的小众物种的基因组从头组装和完善。考虑到小众物种的科研群体较小、可用于全基因组测序的科研经费有限,而基于染色质交互组装的方法成本较低,该方法的应用可节约高密度遗传连锁图谱构建的成本。
5.2 技术的优化、整合和展望
由于基于染色质交互组装基因组的研究尚处于起步阶段,目前仅限于少数模式物种中。因此要充分发挥该方法在基因组组装的作用,需要从以下三个方面着手,进一步优化、整合和完善组装方法。
第一,高分辨率、高质量染色质交互数据的获取。染色质交互数据是该组装方法的基础,其质量的好坏、精度的高低直接制约着基因组组装的准确性和覆盖率。因此,针对特定物种,应该在染色质空间构象捕获实验的准确性、精度等多个层面进行努力。如最近发表在Cell杂志上的通过一种名为原位Hi-C(in situ Hi-C)的方法,测定了人类淋巴母细胞株(GM12878)的全局染色质交互,分辨率高达1 kb[9]。这种原位的方法,使DNA在连接期间仍保留在细胞核内,而不是被释放到溶液中,显著降低了DNA片段随机连接的可能性[9]。
第二,与传统及新兴的大片段文库构建技术、第三代测序相结合,获取高质量的长片段scaffolds。这不仅能提高染色质交互数据的精度,还能提高基因组组装的完整性,同时可减少组装错误。例如,将基于染色质互作的组装方法与大片段文库构建的策略,如双末端测序、最近发展的CPT-Seq(Contiguity preserving transposase sequencing)[24]或新的测序技术(如第三代测序)相结合以获得高质量的组装结果。
第三,与传统遗传图谱信息相结合,相互补充。不管是以物种染色体片段遗传交换为基础的图谱组装法,还是以染色质交互为基础的Hi-C组装法,其都可能存在系统偏好性、甚至错误。因此,在基因组测序工作开展时,可综合两种方法进行基因组组装,实现优势互补,从而获得更加完整准确的参考基因组序列。
[1]https://en. wikipedia. org/wiki/DNA_sequencing. com /.
[2]http://www. genome. gov/sequencingcosts. com /.
[3]Putnam NH, O’Connell B, Stites JC, et al. Chromosomescale shotgun assembly using an in vitro method for long-range linkage[J]. ArXiv, 2015, Available online at:http://arxiv.org/abs/1502. 05331.
[4]Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing:computational challenges and solutions[J]. Nature Reviews Genetics, 2012, 13(1):36-46.
[5]马艳玲, 邓海, 刘中来, 等. 海洋放线菌Streptomyces sp. 大片段DNA基因组文库的构建[J]. 生物技术, 2010(5):1-3.
[6]Claros MG, Bautista R, Guerrero-Fernández D, et al. Why assembling plant genome sequences is so challenging[J]. Biology, 2012, 1(2):439-459.
[7]Dekker J, Rippe K, Dekker M, et al. Capturing chromosome conformation[J]. Science, 2002, 295(5558):1306-1311.
[8]Lieberman-Aiden E, van Berkum NL, Williams L, et al.Comprehensive mapping of long-range interactions reveals folding principles of the human genome[J]. Science, 2009, 326(5950):289-293.
[9]Rao SSP, Huntley MH, Durand NC, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping[J]. Cell, 2014, 159(7):1665-1680.
[10]翟侃, 武治印, 于典科. 染色质构象捕获及其衍生技术[J].生物化学与生物物理进展, 2010, 37(9):939-944.
[11]Dekker J, Marti-Renom MA, Mirny LA. Exploring the threedimensional organization of genomes:interpreting chromatin interaction data[J]. Nat Rev Genet, 2013, 14:390-403.
[12]彭城, 李国亮, 张红雨, 阮一骏. 染色质三维结构重建及其生物学意义[J]. 中国科学:生命科学, 2014, 44(8):794-802.
[13]李国亮, 阮一骏, 谷瑞升, 等. 起航三维基因组学研究[J].科学通报, 2014, 59:1165-1172.
[14]Kaplan N, Dekker J. High-throughput genome scaffolding from in vivo DNA interaction frequency[J]. Nature Biotechnology, 2013,31(12):1143-1147.
[15]Burton JN, Adey A, Patwardhan RP, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions[J]. Nature Biotechnology, 2013, 31(12):1119-1125.
[16]Marie-Nelly H, Marbouty M, Cournac A, et al. High-quality genome(re)assembly using chromosomal contact data[J]. Nature Communications, 2014, 5:5695.
[17]Burton JN, Liachko I, Dunham MJ, et al. Species-Level deconvolution of metagenome assemblies with Hi-C Based contact probability maps[J]. G3:Genes/Genomes/Genetics, 2014, 4(7):1339-1346.
[18]Schneeberger K, Ossowski S, Ott F, et al. Reference-guided assembly of four diverse Arabidopsis thaliana genomes[J]. Proc Natl Acad SciUSA, 2011, 108(25):10249-10254.
[19]Xie T, Zheng JF, Liu S, et al. De novo plant genome assembly based on chromatin interactions:A case study of Arabidopsis thaliana[J]. Molecular Plant, 2015, 8(3):489-492.
[20]Imakaev M, Fudenberg G, McCord RP, et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization[J].Nature Methods, 2012, 9(10):999-1003.
[21]Yaffe E, Tanay A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture[J]. Nature Genetics, 2011, 43(11):1059-1065.
[22]Xie T, Fu LY, Yang QY, et al. Spatial features for Escherichia coli genome organization[J]. BMC Genomics, 2015, 16(1):37.
[23]Dixon JR, Selvaraj S, Yue F, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions[J]. Nature, 2012, 485(7398):376-380.
[24]Adey A, Kitzman JO, Burton JN, et al. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity[J]. Genome Research, 2014, 24(12):2041-2049.
