恶臭假单胞菌HspB的单晶培养及结晶条件优化
2015-09-14邬志杰吴更唐鸿志许平
邬志杰 吴更 唐鸿志 许平
尼古丁是一类广泛存在于土豆、番茄和烟草等茄科植物中的天然生物碱[1],尼古丁对哺乳动物能产生多种致病、致变、致癌效应,且易溶于水,会污染土壤及水环境[2-4]。利用微生物来代谢尼古丁,是治理尼古丁污染的一种有效方法。
恶臭假单胞菌S16是一株尼古丁高效降解菌[5],其利用吡咯途径代谢尼古丁[6]:尼古丁经由N-甲基麦斯明、假氧化尼古丁、3-琥珀酰吡啶、6-羟基-3-琥珀酰吡啶(6-Hydroxy-3-succinoyl-pyridine,HSP)、2,5-二羟基吡啶(2,5-Dihydroxy-pyridine,DHP)、N-甲酰马来酰胺酸、马来酰胺酸、马来酸及富马酸,最终进入三羧酸循环[7,8]。
2011年,单加氧酶HspB及其编码基因hspB被发现,其负责催化HSP转化成DHP[9]。HspB以FAD(黄素腺嘌呤二核苷酸、氧化态)为辅基,以NADH(烟酰胺腺嘌呤二核苷酸,还原态)为辅酶,其催化底物发生羟化机制也已经明确[10]。HspB在整个尼古丁代谢途径中十分关键,将hspB基因缺失后,P. putida S16无法进行尼古丁代谢。因此,获得HspB的结构对于研究其结构与功能的关系十分有利。
通过基因工程构建重组质粒,在大肠杆菌中表达外源蛋白,获得高纯度的蛋白得到单晶,并用于X射线衍射,这是晶体衍射学常用的方法[11]。而前期研究也表明,可以通过大肠杆菌表达纯化得到可溶性的带有His标签的HspB蛋白。然而其晶体的培养却十分困难[12]。虽然His标签对于蛋白结构影响较小[13],但也有文献报道切除His标签可能会有利于晶体的培养,值得尝试[14,15],同时亦有许多蛋白结构是通过切除His标签的蛋白获得的[16-18]。
本研究在前期构建的表达质粒的基础上[12],通过突变PCR定点引入TEV蛋白酶酶切序列,从而实现了其重组蛋白产物能够被TEV蛋白酶切割,达到去除His标签的目的,并利用该蛋白进行结晶研究。旨为后续获得该蛋白结构,阐释其功能和结构间的联系奠定基础。
1 材料与方法
1.1 材料
1.1.1 质粒和菌种 表达质粒pET28a-hspB(抽提自大肠杆菌DH5α),大肠杆菌DH5α 和大肠杆菌BL21(DE3)均为本实验室保存。
1.1.2 主要试剂 FastPfu聚合酶购自全式金公司,Dpn I购自NEB公司,TEV蛋白酶(带有N端6×His标签)为本实验自制,酶质粒小抽试剂盒购自Qiagen公司,实验所用结晶试剂盒购自Hampton Research、EMBio、Jena Bioscience、Molecular Dimension等。其他常用试剂均为国产分析纯。
1.1.3 主要仪器 PCR仪(EDC 810)购自东胜公司,细胞破碎仪购自广州聚能,Ni2+-NTA填充柱料购自Qiagen公司,水平与垂直电泳仪和凝胶成像仪均购自上海天能,ÄKTA prime Plus 购自GE,紫外分光光度计(UV-1800)购自上海美谱达。
1.2 方法
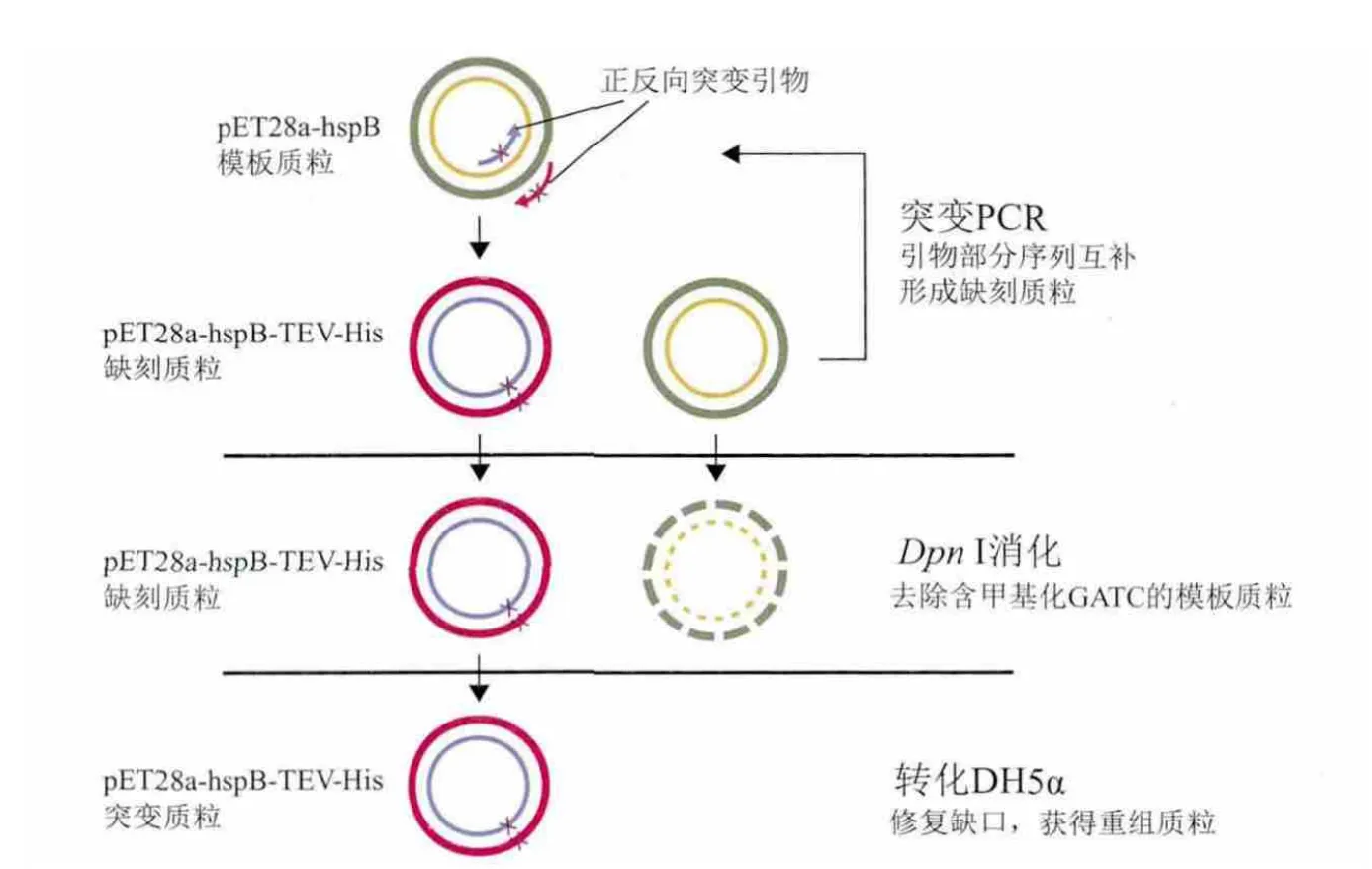
图2 重组质粒pET28a-hspB-TEV-His构建示意图
1.2.1 引物设计、突变PCR及产物鉴定 为实现在重组HspB蛋白C端和6×His标签之间加入TEV蛋白酶酶切识别序列(Glu-Asn-Leu-Tyr-Phe-Gln-Gly)。本研究以表达质粒pET28a-hspB为模板,并由此设计正反向突变引物。P1:5'-GAAAACCTGTATTTTCAGGGCCACCACCACCACCACCAC-3’;P2:5'-GCCCTGAAAATACAGGTTTTCCTCGAGAAAGGTTTCCAT-3'。引物的5'端是TEV蛋白酶酶切识别序列对应的核苷酸碱基序列(下划线部分),3'端则是模板序列(P1的3'端是6×His标签的核苷酸碱基序列;P2的3'端是hspB基因3'端序列(GenBank登录号GQ857548.1)。由生工生物工程(上海)有限公司合成引物。突变PCR反应体系:FastPfu 聚合酶0.5μL,5×FastPfu Buffer 5μL,5×stimulant 5μL,2.5mmo/L dNTP 1.5μL,模板质粒 0.5μL,上下游引物 0.5μL,以 ddH2O补至25μL。反应条件:94℃ 5min;94℃ 15s,55℃ 15s,72℃ 3min,25 个循环 ;72℃10min。产物鉴定:1.0%琼脂糖凝胶电泳检测。
1.2.2 重组质粒pET28a-hspB-TEV-His的构建 向20μL PCR 产物中加入 1μL Dpn I进行消化,37℃,2h。将处理后的PCR产物直接转化E. coli DH5α感受态细胞,筛选阳性克隆。提取质粒送上海美吉生物医药科技有限公司测序。经Blast比对,测序结果正确的质粒命名为pET28a-hspB-TEV-His。
1.2.3 HspB 蛋白的诱导表达 将pET28a-HspBTEV-His转化E. coli BL21(DE3)感受态细胞,于卡那霉素抗性平板上筛选阳性菌落。取阳性单菌落接种于50mL液体LB培养基中,加入终浓度为50mg/mL的卡那霉素,37℃,220r/min培养过夜,作为种子液。以1 100的接种量接种到1 L液体LB培养基中,加入终浓度为50mg/mL的卡那霉素,37℃,220r/min培养至OD600为0.8,加入终浓度为0.2mmol/L的异丙基硫代半乳糖苷(IPTG),16℃,180r/min诱导过夜。4℃,4 200r/min离心收集菌体。以1 L菌对应20mL镍柱平衡液(25mmol/L Tris pH8.0,300mmol/L NaCl,20mmol/L咪唑)的比例重悬,并加入终浓度为100mg/L的抑肽酶、亮抑酶肽及250mg/L的苯甲基磺酰氟,混匀。
1.2.4 镍柱亲和层析 菌液于4℃进行细胞破碎(1300 bar,3次)后,4℃,14000r/min离心,去上清过镍柱两遍。镍柱漂洗液(25mmol/L Tris pH8.0,300mmol/L NaCl,20mmol/L咪唑)漂洗柱子两遍。镍柱洗脱液(25mmol/L Tris pH8.0,300mmol/L NaCl,60mmol/L、200mmol/L咪 唑)5mL洗脱目的蛋白。SDS-PAGE分析。
1.2.5 TEV蛋白酶酶切 按酶与蛋白1 25的质量比加入TEV蛋白酶进行酶切。本研究中采用了两种酶切方法。柱上酶切法:将蛋白上清挂柱后漂洗两次,封闭柱子,然后加入5mL镍柱漂洗液,按比例加入TEV蛋白酶,混匀柱料,4℃过夜。收集漂洗液。透析酶切法:在洗脱液中按比例加入TEV蛋白酶,置于透析袋中,于1 L镍柱漂洗液(25mmol/L Tris pH8.0,300mmol/L NaCl,20mmol/L 咪唑)中 4℃漩涡过夜。
1.2.6 镍柱除杂 在柱酶切法中直接收集酶切后漂洗液即可;透析酶切法则需要将透析液流穿镍柱,收集流出液及漂洗液。两种方法都需要用500mmol/L咪唑洗脱液洗脱柱上没有酶切完全的蛋白和TEV酶,并进行SDS-PAGE检测。
1.2.7 凝胶过滤层析 预先用平衡缓冲液(25mmol/L Tris pH8.0,300mmol/L NaCl,2mmol/L DTT,1mmol/L EDTA)平衡 Superdex200 HiLoad 16/600 凝胶层析柱(流速1.0mL/min,压强上限0.4 MPa)至电导曲线平直。将收集的流出液及漂洗液注入5mL上样环中。用一个柱体积(约130mL)的平衡缓冲液洗脱并收集洗脱液。结合紫外吸收峰和SDSPAGE分析纯化效果。4℃,4 500r/min离心浓缩蛋白溶液,Bradford法测定蛋白浓度至12mg/mL,直接进行点晶或使用液氮速冻,并于-80℃保存。
1.2.8 SDS-PAGE分析 采用12%分离胶和5%浓缩胶制胶,电泳后使用考马斯亮蓝R250染色。
1.2.9 点晶 蛋白冰上解冻后,4℃,14000r/min离心5min后点晶。本实验采用悬滴扩散法进行结晶。使用48孔(24孔)结晶板,下槽孔中的结晶试剂为 80μL(160μL)。在硅化玻片上依次滴加 1μL 蛋白溶液和1μL结晶试剂,再将盖玻片反扣覆盖在池孔上并用凡士林密封空隙。恒温静置培养。定期在显微镜下观察晶体生长情况。
1.2.10 结晶条件优化 以Index 2试剂盒的第83个条件为基础,对结晶条件做进一步优化。优化蛋白浓度:尝试13.5、12、9.5和7.5mg/mL浓度蛋白进行点晶。设计正交实验对pH、沉淀剂和盐离子浓度进行优化:沉淀剂的浓度设置了以1%为单位,尝试了12个梯度;盐离子浓度以0.1mol/L为单位,尝试了12 个梯度;pH以0.1为梯度尝试了10个梯度;同时选择了不同的沉淀剂(PEG200-10000)、盐(各类镁盐及盐酸盐)和pH缓冲液成分(Mes、Bis-Tris和HEPES),确定最优生长条件。添加剂筛选:控制原结晶条件不变,在液滴中加入0.2μL添加剂(96种添加剂均来自Additive Screen试剂盒)后静置培养。优化培养温度:尝试4、8、14、18和20℃下培养晶体。最后也尝试了微接种法,以获得单晶。
2 结果
2.1 pET28a-hspB-TEV-His重组质粒的构建与鉴定
突变PCR反应后,扩增产物由琼脂糖凝胶电泳鉴定,在4-8 kb间可见一条与pET28a-hspB-TEVHis质粒理论大小(6.4 kb)相符的条带(图3)。该PCR产物中包含环装缺刻质粒(目的产物)和痕量模板质粒,经Dpn I消化后转化DH5α,挑取单菌落送测序,经比对,序列完全正确。

图3 pET28a-hspB-TEV-His重组质粒构建
2.2 HspB-TEV-His蛋白的镍柱亲和层析
菌体破碎后,离心收集上清,通过镍柱亲和层析纯化,经SDS-PAGE检测,在45kD处有目的蛋白条带(图4),与预期(44.6kD)一致。上清、洗脱液中均有目的蛋白,60mmol/L咪唑下可见少许杂带(66kD处),判断可在之后的凝胶过滤层析中去除,故合并洗脱液。可见,该重组蛋白表达高、可溶性好且杂蛋白较少。
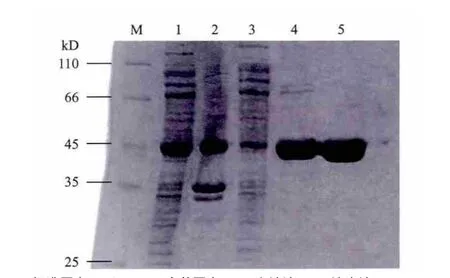
图4 HspB-TEV-His蛋白的镍柱亲和层析
2.3 TEV蛋白酶酶切效果比较
柱上酶切法 经SDS-PAGE分析(图5),漂洗液中只有切除His标签的HspB(45kD),洗脱液中则存在酶切不完全仍带有His标签的HspB-TEV-His(45kD)和TEV蛋白酶(27kD)。
透析酶切法 经SDS-PAGE分析(图5),流出液中有切除His标签的HspB蛋白和少量TEV蛋白酶,洗脱液中有未切开的HspB和TEV蛋白酶。
两种酶切方法均可切除His标签,且保证较高的纯度。对比两者洗脱液,显然在柱酶切中未切开的蛋白比透析酶切更多,说明透析酶切效率更高。

图5 TEV蛋白酶酶切效果比较
2.4 HspB的凝胶过滤层析
Superdex200柱层析紫外吸收峰单一,峰型对称陡峭,位置在56管(78.4mL)处,此处对应于44kD,大小正确(图6-A),取对应峰位置的蛋白进行SDS-PAGE分析发现,纯化的蛋白为单一条带(图6-B),达到晶体初筛所要求的纯度。收集峰尖位置左右的5管蛋白进行浓缩。

图6 HspB的凝胶过滤层析
2.5 HspB蛋白结晶条件初筛
经过大量的初筛条件筛选后,在Index 2试剂盒(Hampton Research)的第83个条件下有晶体生长,条件为25% PEG3350,0.1mol/L Bis-Tris pH6.5,0.2mol/L MgCl2,14℃。晶体呈薄片状,且有孪晶现象(图7-A),不适合X-射线衍射数据收集,需要做进一步的优化工作。
2.6 HspB蛋白结晶条件优化
确定的最优结晶条件是蛋白浓度7.5mg/mL,22% PEG3350,0.1mmol/L Bis-Tris pH6.5,0.21mol/L MgCl2,18℃,1 50的比例加入晶种,蛋白溶液与下槽液体积比为2 1的条件下,能够获得立体的块状单晶(图7-B),该晶体在上海光源进行X射线衍射后发现分辨率达到了1.8 Å。
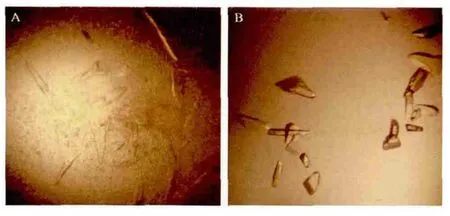
图7 HspB晶体的优化
3 讨论
本研究所采用经典的QuickChange定点突变方法[19,20](图 2):设计一对包含突变位点的引物,和模版质粒退火后用Pfu聚合酶“循环延伸”(聚合酶按照模版质粒延伸引物,一圈后回到引物5'端终止,再经过反复加热退火延伸的循环)。正反向引物的延伸产物退火后配对成为带缺刻的开环质粒。Dpn I酶切延伸产物,由于原来的模版质粒来源于常规大肠杆菌,是经dam甲基化修饰的,对Dpn I敏感而被切碎(Dpn I识别序列为甲基化的GATC,GATC在几乎各种质粒中都会出现,而且不止一次),而体外合成的带突变序列的质粒因无甲基化而未被切开,因此得以成功转化(大肠杆菌体内修复缺刻质粒),即可得到突变质粒的克隆[21]。前期实验构建了pET28a-HspB质粒,表达纯化得到HspB-His蛋白,用于结晶研究。由于晶体质量较差,不能用于衍射[12]。蛋白C端序列存在超过30个氨基酸是非折叠区域(Foldindex预测),而这些氨基酸所形成的柔性肽段,会导致蛋白在构象上的不均一,阻碍晶体的生长[15]。然而完全或部分去除这些序列会导致蛋白不表达或沉淀(数据未显示),暗示着该序列对于HspB结构有着稳定作用。因此本研究选择去除其C端的His标签(6×His短肽也是柔性肽),以提升蛋白晶体的质量。
前期实验尝试插入凝血酶酶切序列,然后发现其无法被凝血酶切开。推测是由于其识别序列(Leu-Val-Pro-Arg-Gly-Ser)较短,仅6个氨基酸,且前3个氨基酸(Leu、Val和Pro)均是非极性氨基酸,在表达时可能被折叠进了蛋白疏水核,没有暴露在水环境中[22];或者是由于其酶切位点在Arg和Gly间,由于空间位阻,导致无法切除目的氨基酸[23]。
本研究总结了前期实验的经验,选择了TEV蛋白酶作为工具酶,其主要考量在于:(1)其识别序列(Glu-Asn-Leu-Tyr-Phe-Gln-Gly)更长,为7个氨基酸,且除了Phe疏水外,其余氨基酸皆亲水,不易被折叠进疏水核内;(2)其酶切位点位于Gln和Gly间,离蛋白更远,会减小空间位阻的影响。
TEV蛋白酶酶切时,需要解决了3个问题:(1)新增的步骤带来的蛋白损失问题;(2)保持蛋白纯度和性质的问题;(3)纯化路线的可操作性和时间问题。因此,本研究比较了柱上酶切和透析酶切的效率及纯化效果发现,虽然在柱酶切比较省时省力,然而酶切效率较低,这可能是由于TEV蛋白酶与底物HspB蛋白都带有His标签,因而结合在柱料上,发生的是固固反应,分子间接触几率不如溶液反应那么高,并且酶固定化后会降低酶活[24];而透析酶切是溶液反应,在酶切的同时进行咪唑浓度的稀释,可以节省时间,方便下一步的镍柱除杂。因此最后选择了透析酶切的策略。
蛋白的均一性是由镍柱除杂和凝胶过滤层析来保证的。镍柱除去了未切开的HspB蛋白,保证产物中仅有不带标签的HspB蛋白,这是无法通过凝胶过滤层析实现的。均一性对于蛋白结晶的影响是很大的,因此为了保持最大均一性,只取凝胶过滤层析中紫外吸收峰半峰对应的5管蛋白进行浓缩点晶,而非整个紫外吸收峰对应的蛋白[25]。这是由于均一性越高,其蛋白的大小形状也越接近,在凝胶过滤层析中,也就越倾向于在同一位置出峰。
因此最终确定下来的纯化路线能够获得纯度高、较均一的HspB蛋白,并且没有降解存在。同时整个纯化过程都在4℃进行,在最终保存液中也添加了二硫苏糖醇以保护蛋白。
培养得到的单晶在上海光源进行X射线衍射后发现,虽然分辨率达到了1.8 Å,但是在后续处理数据时发现其仍有孪晶趋势,这可能是由于TEV蛋白酶酶切后留下的肽(Glu-Asn-Leu-Tyr-Phe-Gln)具有一定的柔性所致,相比6×His标签,其在结晶条件(pH6.5)下带电性更少,因而减少了同性相斥的现象,更利于蛋白分子有序堆积[26]。因此接下来将继续优化C端序列,进一步提高其构象均一性。底物的存在有利于晶体的生长[27],因此也将尝试进行与底物的共结晶,进一步优化晶体。HspB是恶臭假单胞菌尼古丁代谢通路中的关键酶,该途径中其他酶的结构多已见诸报道[28],而HspB的结构仍旧未知。了解HspB结构不仅有助于理解其功能和结构之间的联系,完善整个吡咯途径,还对定向进化提高酶活及进行工业化应用具有积极的指导意义。
4 结论
本研究以恶臭假单胞菌HspB表达质粒pET28a-HspB为模板,通过PCR插入TEV蛋白酶酶切序列,测序验证重组质粒pET28a-HspB-TEV-His。选用大肠杆菌BL21(DE3)进行表达,经过镍柱亲和层析、TEV蛋白酶酶切、镍柱除杂以及凝胶过滤层析获得高纯度蛋白。
优化结晶条件后,确定了单晶培养条件是蛋白浓 度 7.5mg/mL,22%PEG3350、0.1mol/L Bis-Tris pH6.5、0.21mol/L MgCl2,18℃,以1 50的比例加晶种,蛋白溶液与下槽液体积比为2 1的条件;单晶的分辨率达到1.8 Å。
[1]Doolittle DJ, Winegar R, Lee JK, et al. The genotoxic potential of nicotine and its major metabolites[J]. Mutat Res, 1995, 344:95-102.
[2]Campain JA. Nicotine:potentially a multifunctional carcinogen?[J]. Toxicological Sciences, 2004, 79(1):1-3.
[3]Hecht SS, Hochalter JB, Villalta PW, et al. 2’-Hydroxylation of nicotine by cytochrome P450 2A6 and human liver microsomes:formation of a lung carcinogen precursor[J]. Proceeding of the National Academy of SciencesUSA, 2000, 97(23):12493-12497.
[4]Novotny TE, Zhao F. Consumption and production waste:another externality of tobacco use[J]. Tob Control, 1999, 8(1):75-80.
[5]Wang SN, Xu P, Tang HZ, et al. Biodegradation and detoxification of nicotine in tobacco solid waste by a Pseudomonas sp. [J].Biotechnology Letters, 2004, 26(19):1493-1496.
[6]Wang S, Liu Z, Tang H, et al. Characterization of environm-entally friendly nicotine degradation by Pseudomonas putida biotype A strain S16. [J]. Microbiol, 2007, 153:1556-1565.
[7]Wang SN, Xu P, Tang HZ, et al. ‘Green’ route to 6-hydroxy-3-succinoyl-pyridine from(S)-nicotine of tobacco waste by whole cells of a Pseudomonas sp. [J]. Environmental Science Technology, 2005,39:6877-6880.
[8]Tang H, Wang L, Wang W, et al. Systematic unraveling of the unsolved pathway of nicotine degradation in Pseudomonas[J].PLoS Genet,2013, 9(10):e1003923.
[9]Tang H, Wang S, Ma L, et al. A novel gene, encoding 185 6-hydroxy-3-succinoylpyridine hydroxylase, involved in nicotine degradation by Pseudomonas putida strain S16. [J]Appl Environ Microbiol, 2008,74(5):1567-1574.
[10]Tang H, Yao Y, Zhang D, et al. A novel NADH-dependent and FAD-containing hydroxylase is crucial for nicotine degradation by Pseudomonas putida[J]. J Biol Chem, 2011, 286:39179-39187.
[11]McPherson A. Crystallization of biological macromolecules[M].1st ed. New York:Cold Spring Harbor Laboratory Press, 1999.
[12]胡传明, 于浩, 唐鸿志, 等. 6-羟基-3-琥珀酰吡啶单加氧酶的纯化与结晶条件[J]. 微生物学通报, 2014, 41(9):1779-1784.
[13]Carson M, Johnson DH, McDonald H, et al. His-tag impact on structure[J]. Acta Cryst D, 2007, 63:295-301.
[14]Bergfors TM.Protein crystallization:techniques, strategies, and tips:a laboratory manual[M]. California:InternationalUniversity Line, 1999.
[15]Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization[J]. Acta Cryst D, 2006, 62:116-124.
[16]Strong M, Sawaya MR, Wang S, et al. Toward the structural genomics of complexes:crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis[J]. Proceedings of the National Academy of Sciences, 2006, 103(21):8060-8065.
[17]Hall TMT, Porter JA, Young KE, et al. Crystal structure of a Hedgehog autoprocessing domain:homology between Hedgehog and self-splicing proteins[J]. Cell, 1997, 91(1):85-97.
[18]Bauman JD, Das K, Ho WC, et al. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design[J]. Nucleic Acids Research, 2008, 36(15):5083-5092.
[19]Zheng L, BaumannU, Reymond JL. An efficient one-step sitedirected and site-saturation mutagenesis protocol[J]. Nucleic Acids Research, 2004, 32(14):e115.
[20]Li J, Li C, Xiao W, et al. Site-directed mutagenesis by combination of homologous recombination and DpnI digestion of the plasmid template in Escherichia coli[J]. Analytical Biochemistry, 2008,373(2):389-391.
[21]Carter P. Site-directed mutagenesis[J]. Biochemical Journal,1986, 237(1):1.
[22]Brondyk WH. Selecting an appropriate method for expressing a recombinant protein[J]. Methods Enzymol, 2009, 463:131-147.
[23]Estell DA, Graycar TP, Miller JV, et al. Probing steric and hydrophobic effects on enzyme-substrate interactions by protein engineering[J]. Science, 1986, 233(4764):659-663.
[24]Mateo C, Palomo JM, Fernandez-Lorente G, et al. Improvement of enzyme activity, stability and selectivity via immobilization techniques[J]. Enzyme Microb Technol, 2007, 40:1451-1463.
[25]Rengachari S, Aschauer P, Sturm C, et al. Purification,crystallization and preliminary X-ray diffraction analysis of a soluble variant of the monoglyceride lipase Yju3p from the yeast Saccharomyces cerevisiae[J]. Acta Crystallographica Section F :Structural Biology Communications, 2015, 71(2):242-245.
[26]Kantardjieff KA, Rupp B. Protein isoelectric point as a predictor for increased crystallization screening efficiency[J]. Bioinformatics,2004, 20(14):2162-2168.
[27]Smits SHJ, Mueller A, Grieshaber MK, et al. Coenzyme-and Histag-induced crystallization of octopine dehydrogenase[J]. Acta Crystallogr F:Struct Biol Crystallization Commun, 2008, 64(9):836-839.
[28]Chen D, Tang H, Lv Y, et al. Structural and computational studies of the maleate isomerase from Pseudomonas putida S16 reveal a breathing motion wrapping the substrate inside[J]. Molecular Microbiology, 2013, 87(6):1237-1244.
