环境微生物宏基因组学数据库利用
2015-10-28王慧丽郭安源
王慧丽郭安源
(1.文华学院城市建设工程学部环境工程系,武汉 430074;2.华中科技大学生命科学与技术学院,武汉 430074)
环境微生物宏基因组学数据库利用
王慧丽1郭安源2
(1.文华学院城市建设工程学部环境工程系,武汉 430074;2.华中科技大学生命科学与技术学院,武汉 430074)
宏基因组学技术产生的数据是研究环境微生物的宝贵资源,国际上已有微生物计划、海洋计划、生命普查等大项目,采集和测序的样本量数以百万计,产生了海量的环境宏基因组学数据,并以此建立了几十个相关宏基因组数据库和平台。主要从以下几个方面综述环境宏基因组学的研究进展和已有资源:环境宏基因组学国际合作大项目、宏基因组学数据库和宏基因组学数据在线分析平台。将结合相应的数据库网站介绍其项目详情、样本来源、数据类型、使用方式和分析结果等,以便研究者全面了解此类数据并能快速找到和利用相关资源。
宏基因组学;数据库;资源;微生物
微生物在物质合成、降解、碳氮元素循环等方面具有十分重要的生态功能[1]。微生物的种类和数量繁多,且大多数难以单独分离、培养和鉴定。近年来,高通量二代测序技术的发展给环境微生物的研究带来了新的方法和策略,其在微生物基因组中的应用产生了宏基因组学(Metagenome)这一新的学科,为地球生物资源的普查和鉴定提供了新的技术和方法。
宏基因组这一概念由Handelsman[2]于1998年提出,其含义是将环境中全部微生物的遗传信息看作一个整体,对环境样品中细菌和真菌的基因组总和进行研究。宏基因组学研究方法避开了微生物需要培养提取的过程,还可以揭示微生物之间及其与环境之间相互作用的规律[1]。宏基因组学研究已经成为微生物研究的热点和前沿,在环境生物多样性的探测、气候变化、极端环境、人体肠道、石油污染修复、生物冶金等领域,取得了一系列重要成果[3]。国际上多个不同组织和研究联盟对全球的不同环境进行微生物多样性检测和探查。这些研究采集和产生了大量的不同环境微生物的数据,基于此,目前国际上建立了几十个有关环境生物资源和宏基因组学数据的大数据库。基于作者对生物数据库开发和使用方面的深入研究和体会,结合环境微生物学研究需求,本文着重介绍国际上大型的环境宏基因组学项目、环境宏基因组学相关数据库和分析平台,以便相关人员更好的利用这些资源。
1 环境宏基因组学项目介绍
宏基因组学技术实现以来,国际上针对全球的环境微生物普查实施了几大项目计划,主要有地球微生物组计划(The Earth Microbiome Project,EMP)、全球海洋采样(Global Ocean Sampling,GOS)和海洋生命普查(Census of Marine Life,CoML)。
1.1 地球微生物组计划(EMP)
地球微生物组计划(The Earth Microbiome Projectm,EMP)[4]是一个分析全球范围内微生物群落、描述全球微生物多样性和功能的项目。其目标是使用宏基因组、宏转录组和扩增子测序分析近20万个来自不同地球环境和生态系统的样本,产生了全球基因图谱描述每个生物群落的蛋白质和环境代谢模型,以及大约50万个重建的微生物基因组,建立了全球代谢模型和数据可视化分析的门户网站。
EMP项目主要目标和任务:(1)基因地图集(Gene Atlas),研究中获得的所有信息的集中存储库,提供可搜索的格式存储所有的序列、注释信息和环境元数据。(2)地球微生物组的基因组(Earth microbiome assembled genomes),包含所有EMP数据中组装的基因组,并且使用自动注释流程进行注释和分析。(3)地球微生物组可视化接口(Earth Micro-biome Visualization Portal),建立交互式可视化软件处理数据,使之方便易用。人们能从微生物空间的角度查看地球,描述环境和基因组功能,以便整合EMP数据发现新的生态理论。(4)地球微生物组代谢重建(Earth microbiome metabolic reconstruction),基于宏基因组代谢组数据描述和软件预测,描述代谢物随时间和生物地理空间的改变。
EMP项目的数据存放在http://qiita.microbio. me/网站,这是一个完全开源的微生物组数据存储和分析资源,建立在广泛使用的QIIME包之上,可用于分析组学数据。目前Qiita网站中存储有来自158个研究项目的33 285个样本的数据,需要注册使用。注册用户登录后,可以通过“Study”菜单下的“View Studies”子菜单来搜索已有的数据,如搜索“Earth Microbiome Project”可以得到该项目的所有数据,然后可以选择相应的数据点击“Add to Analysis”进行分析,可选择的分析有稀释度和物种多样性等。
1.2 全球海洋采样(GOS)
全球海洋采样(Global ocean sampling,GOS)是一个探测海洋生物基因组的项目,它的目标是评估海洋微生物群落的遗传多样性,了解它们在自然的基本进程中扮演的角色。J. Craig Venter研究所(JCVI)的科学家们自2003年开始对全球范围内的海洋进行采样,目的是通过对生活在这些水域中的微生物的DNA采样、测序和分析以了解无尽海洋的奥秘。2003-2008年,采样主要集中在美国西海岸,同时与其他合作者在一些极端环境进行了采样。随后于2009-2010年沿墨西哥海岸进行采样,然后离开美洲进发到欧洲,对波罗的海、地中海和红海水域的微生物进行采样研究分析。对Sargasso海采样测序的宏基因组数据分析,鉴定了1 800个独特的基因组、148个之前研究未涉及的细菌种群和120万个未报导的新基因[5]。该研究证明了基因组测序方法鉴定宏基因组的可行性,并为海洋微生物数据增添了许多资源和技术。为了分析这些数据,科学家们还开发了一系列新的生物信息学分析方法和软件,促进了多个相关学科的发展。
全球海洋采样项目的数据同时也提交到了NCBI的SRA数据库和高级海洋微生物生态研究和分析领域基础设施CAMERA,CAMERA是一个在线海洋宏基因资源库。
1.3 海洋生命普查(CoML)
海洋生命普查(Census of Marine Life,CoML)发起的目的是评估和解释全世界海洋生命的多样性、分布和丰富性,以及它们的过去、现在和未来。海洋生命普查(2000-2010年)已经成为一个全球协作的科学项目,有超过80个国家的研究者参与,是世界上第一个全面的海洋生命普查,结果于2010年在伦敦发布。大部分研究报道发表在PLoS One杂志,并且有一个专门的文章合集(http://www.ploscollec tions.org/static/comlCollections.action)。海洋生命普查项目包含14个野外的普查项目和4个非野外项目(表1)。
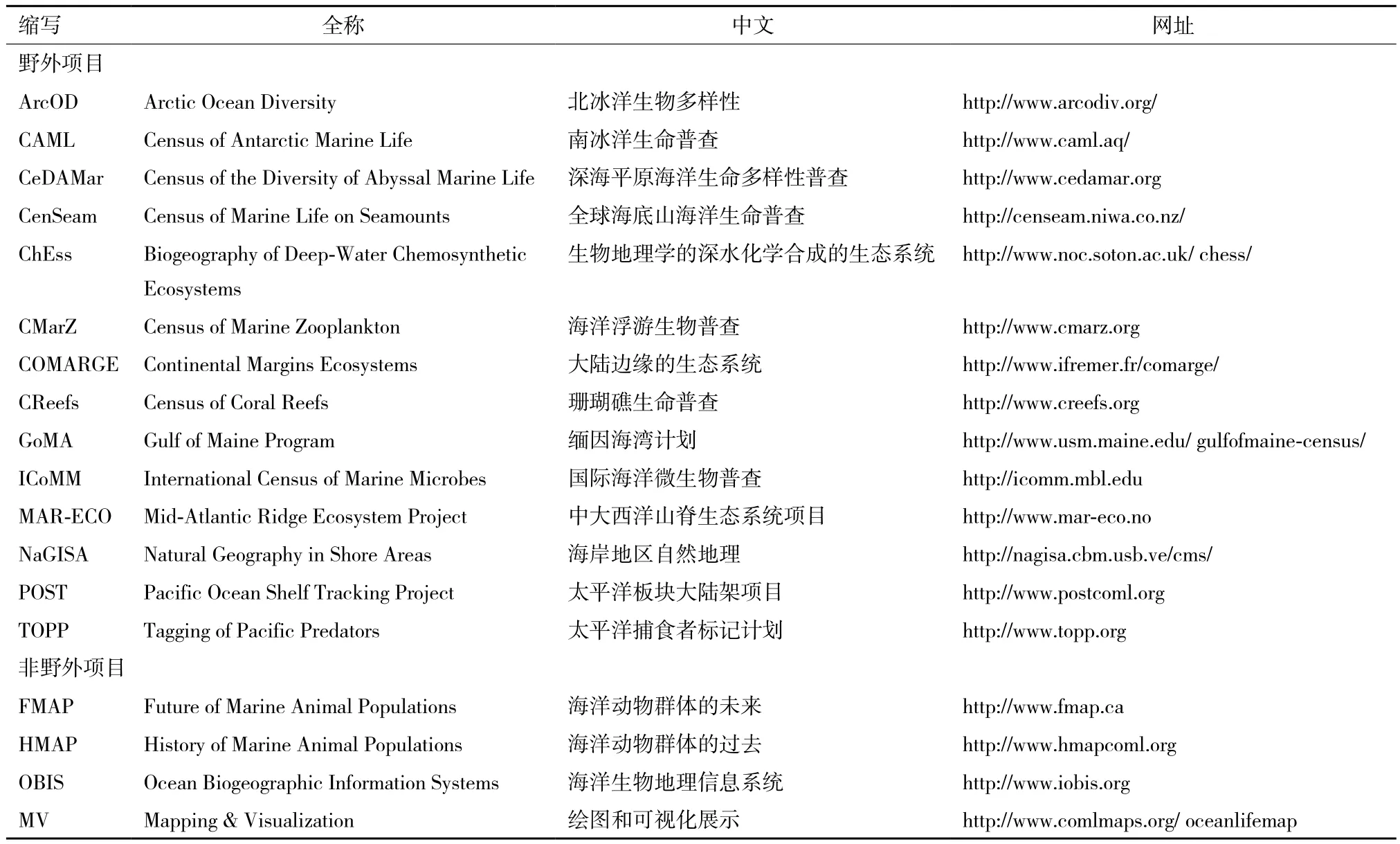
表1 海洋生命普查项目包含的子项目信息
该普查同时形成了一个名为“First Census of Marine Life 2010:Highlights of a Decade of Discovery”(2010年第一次海洋生命普查:10年发现的亮点)的64页的报告[6],描述海洋生命普查中10年探索、研究和分析发现的科学亮点。http://www.coml. org/census-resources网站列出了此普查项目的一些资源列表,对表1的各个项目都有网站详细描述其成果。同时该项目主要的参与国家和地区也分别有各自的网站介绍本国参与和获得的成果,包括澳大利亚、加拿大、加勒比海、中国、欧洲、印度洋和美国。
2 环境宏基因组学数据库
上述EMP、GOS和CoML三个国际环境宏基因组大项目获得了大量的样本和数据,也分别建立了相应的数据库存储数据。此外,国际上还有其他一些专业数据库存储环境微生物宏基因组学数据,供全世界的研究者使用,以下分别介绍之(表2)。
2.1 基因组在线数据库(GOLD)
基因组在线数据库(Genomes Online Database,GOLD)[7],是一个存储基因组和宏基因组测序项目信息及其元数据(Metadata)的网络资源。GOLD中包含的研究项目有2万多个,其中546个是宏基因组研究项目。这些宏基因组样本的来源使用了谷歌地图和谷歌地球来展示,遍布全世界各地,如美国、澳大利亚、新西兰、巴拿马、马来西亚等,样本来源环境包括温泉、淡水、海洋、土壤、绿色肥料、人和动物身上的微生物群落等。GOLD包含的生物样本中环境相关的样本有6 777个。GOLD数据库提供了3种检索方式,快速搜索、高级搜索和元数据搜索。其中快速搜索允许用户使用最常用的域或关键字检索数据库。高级检索则可以对元数据的各个域和数据库中各个分类层次进行精确的查找。元数据搜索的目的是使用各种元数据标识符查询数据库。各种搜索选项卡包含图形和表格表示其描述的项目或生物的数量,以便获得一个项目和样品的整体情况,同时可根据选择标准产生一个可排序的表和饼形图方便参考。
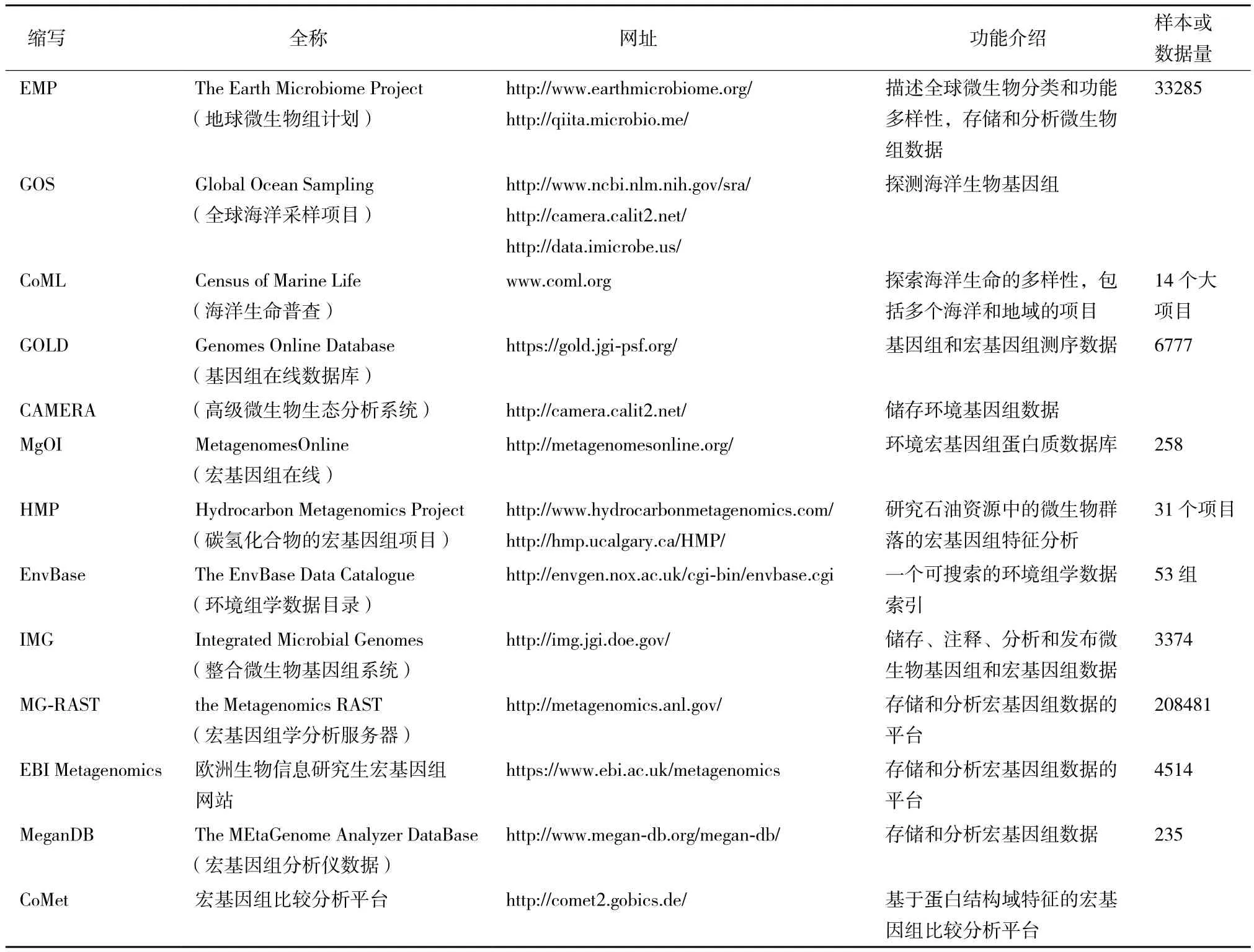
表2 国际上已有环境微生物宏基因组学数据库资源列表
2.2 CAMERA
CAMERA[8]提供了很多重要的资源,包括质量可靠的经过校验的环境基因组数据库、用户提交和保存环境相关分子序列数据的平台,以及开放的计算资源用于宏基因组比较分析等,特别是CAMERA的计算资源,包括大规模的BLAST计算能力和其他流程化分析能力。CAMERA项目受到Gordon和Betty Moore 基金会(GBMF)的海洋微生物学计划(Marine Microbiology Initiative)和美国国家自然科学基金委资助,主要服务于海洋微生物学领域和其他用户。但是由于GBMF基金会资助的终止,CAMERA从2014年7月1日起不再为科学领域提供计算需求,即不再接受新的计算申请提交。但是CAMERA还将继续维护其收集的大量校验好的数据,并提供免费的开放获取服务,这是通过CAMERA的数据分发中心来完成的,同时可以链接到海洋微生物真核转录组测序项目。其数据分发中心提供下载的数据包括:宏基因组和基因组数据,该项目用到的参考基因组和蛋白质序列,约750个来自数百个不同物种样本的转录组测序数据,以及微生物基因组测序计划项目(The Microbial Genome Sequencing Project)中获得的海洋环境宏基因组和生态基因组及其比较分析的数据。这些海洋环境宏基因组和基因组测序筛选的样本主要是来自海洋和开放沿海的细菌浮游生物群落。该项目是JCVI研究所完成的,其完成了177个海洋微生物的测序、组装和自动注释。这些物种的生理多样性较好,包括固碳、光能自养型、光能异养型、硝化细菌和甲烷氧化菌等。PLoS Biology杂志于2007年5月专门为此项目做了一期题为 Ocean Metagenomics Collection的专刊[9]。
2.3 MetagenomesOnline(MgOI)
MgOI是一个校准过的环境宏基因组蛋白质数据库,包括病毒和微生物鸟枪法测序的宏基因组中的预测蛋白质及其注释资源。MgOI中的样本信息比较丰富,包括样品来源、地理描述、环境参数、取样和制备方法及环境本体论条目(Environmental ontology terms)等,并且这些样本进一步被用MgOI的样本描述方法归类,使之容易被理解和比较。MgOI最初是被设计为VIROME宏基因组项目[10]注释流程中的环境蛋白质数据库。用户可以在网站上使用MgOI BLAST工具对数据库进行小规模(小于10条序列)的同源序列搜索,或者下载全部的数据库进行本地化的搜索。目前,MgOI数据库中包括50个项目的总共258个文库(包括病毒、原核和真核)的数据,其中总蛋白质条数为56 254 299,总氨基酸数目达6 480 011 292。
2.4 Hydrocarbon Metagenomics Project(HMP)
为了应对碳氢化合物相关能源研究,加拿大启动了碳氢化合物的宏基因组项目(HMP)[11]。这个项目研究加拿大石油资源中的微生物群落的宏基因组特征,目的是开发新的和改进现有的生物过程,提高碳氢化合物的获取,减少能源使用和温室气体排放。这个项目产生了大量序列和分析数据,同时还开发了宏基因组分析工具和流程。HMP数据库中含有来自31个项目的宏基因组数据,主要样本来自含油砂、油田、尾矿池、煤层等环境。宏基因组数据包括采用454或Illumina测序得到的原始或经过质控的测序数据,经过序列组装的contig结果等。每个宏基因组样本,都有一个单独的页面可以查看或者下载原始的和处理后的数据,以及每一步的分析结果。这些项目数据绝大部分都上传到IMG/M、MG-RAST或NCBI SRA数据库中,并提供了其链接。
2.5 The EnvBase Data Catalogue
EnvBase是一个可搜索的环境组学数据索引,属于英国国家环境研究委员会(National Environment Research Council,简称NERC)下的环境生物信息中心(NERC Environmental Bioinformatics Centre,简称NEBC)。EnvBase包括不同研究组提交的53组环境组学数据集。
2.6 IMG Data Management
这是美国能源部联合基因组研究所(DOE's Joint Genome Institute,简称JGI)的整合微生物基因组系统(Integrated Microbial Genomes,简称IMG),该系统的目标是注释、分析和发布JGI测序的微生物基因组和宏基因组数据,因此同时含有宏基因组数据库和分析平台。只要同意其数据发布政策,IMG面向全世界的科学家提供免费的宏基因组数据注释、分析和整合的支持,同时也可以自由获取整合的基因组和宏基因组的比较分析。截至2014年12月31日,IMG有来自6大洲,88个国家的10 310名用户。IMG系统的数据分发政策是上传的基因组和宏基因组数据自从这些数据可以用于分析时起保持私有状态2年,之后将会公开给全世界的科研界共享。到2015年初,IMG共有来自所有生命界的32 802个基因组数据集和5 234个宏基因组数据集,其中IMG宏基因组数据仓库(IMG/M)中包含245个项目的3 374个公开的宏基因组数据集,对应3 161个独立样本,其中环境样本数据集有2 021个(空气31个,水环境1 207个,陆地环境783个),其他的为工程领域和宿主相关的样本[12]。用户可以免费注册一个账号,登录后可以浏览查看和分析数据库中已有的公共数据,也可以上传自己的数据进行分析。IMG数据中,20%的基因组和75%的宏基因组数据样本是JGI测序的。
IMG中的基因使用了多个功能资源进行鉴定和注释,这些资源包括COG、KOG、KEGG、PFAM、TIGRfam、MetaCyc和Gene Ontology。IMG数据统计页面含有当前IMG中的基本数据统计,包括基因组统计(如处于完成和草图状态的细菌、古细菌、真核、质粒、病毒、微生物组和基因组片段的数目)、基因统计(各类蛋白质编码基因和RNA编码基因的统计以及这些基因在COG、KOG、Pfam等数据库中的注释比例)、功能统计(有Gene ontology和KEGG pathway功能注释的基因统计、各种功能酶统计等)和组学实验统计(蛋白质实验、转录组研究、甲基化实验和必需基因实验等)。其中基因组统计中的微生物组即为宏基因组(图1-A)。点击微生物组下面的数字或者首页左边的统计信息中的Metagenome后,进入“Find Genomes”页面,也即“Genome Browser”页面,此页面使用分页方式展示了所有公开的宏基因组研究项目信息,也可以使用关键字进行过滤筛选(图1-B)。然后选择其中一个数据集点击其中的“Genome Name/Sample Name”可以进入该数据集的具体信息页面,包括该研究名称、样本名、采集时间、地点等基本信息,同时也包含该数据集的基本统计信息和基因信息。此页面中的功能还包括把该数据加入待分析的购物车、浏览已分析好的基因组、BLAST分析基因组和下载数据(图1-C)。此页面的数据统计信息包括宏基因组数据统计,如序列数目、碱基数目、G+C含量、基因数目等(图1-D),也可以根据关键字查询相关注释基因信息,浏览基因组组装的scaffold和浏览基因长度分布信息等(图1-E)以及浏览基因组在进化和系统发生学上的分布(图1-F)。
3 环境宏基因组数据分析平台
随着宏基因组技术产生的大量数据需要分析,很多研究组发展了宏基因组学数据分析方法和工具。这些工具在用户使用上可以分为两类:一类是可以独立下载安装的宏基因组分析软件包,使用它一般需要较强的生物信息学基础;另一类是在线的宏基因组数据分析平台,这是为了使生物信息基础薄弱的研究者也能分析宏基因组数据而开发的,因此较为简单易用。本文着重介绍这一类在线的宏基因组分析平台。这类平台可以上传和保存数据,故通常也包含有存储宏基因组数据的功能,即它们通常既是数据库也是分析平台。如上文提到的IMG整合微生物基因组系统,该系统既可以分析IMG数据库中存储的公共数据,也可以上传新的数据进行分析。下面再介绍其他几个比较常见的宏基因组数据在线分析平台。
3.1 MG-RAST(the Metagenomics Analysis Server)
MG-RAST服务器是一个基于序列数据提供微生物群落的定量分析的宏基因组自动分析平台[13],这个服务器主要提供数据上传、质量控制、自动注释和分析原核宏基因组鸟枪法测序数据。MGRAST启动于2007年,有超过12 000个注册用户。截至2015年9月,MG-RAST服务器含有208 481个宏基因组数据集,共83.42 Tb碱基数据,其中近3万个宏基因组数据是公开的。对这些公开的数据,可以直接下载、分析和查询注释信息,如http://metagenomics.anl.gov/metagenomics.cgi?page=Ana lysis&metagenome=4440039.3是其中一个公开数据的分析页面,用户可以选择不同的数据展示方式(如表格、树图、热图、柱图等)进行分析和注释,也可以查看某个宏基因组数据的详细信息如页面http:// metagenomics.anl.gov/?page=MetagenomeOverview&met agenome=4440036.3。
MG-RAST是目前使用最为广泛的宏基因组数据在线分析服务器,目前更新到第3版本。注册用户可以上传宏基因组数据(测序的fastq格式即可)进行分析。上传数据后,可以使用其managebox工具提供的join paired-ends 功能把双端测序的两个文件整合成一个文件便于后续分析。接下来进行参数选择,有一些对数据过滤的选项(如低质量序列过滤和宿主物种序列过滤),可以根据具体情况选择,或者使用默认参数。分析完成后,MG-RAST将对数据结果以多种形式展现,如图2所示。这些分析包括与各种注释数据库的匹配序列数目和相似性匹配程度(图2-A)、各类蛋白质在数据库中的注释类型统计(图2-B)、Subsystems系统注释[14]的蛋白质分类和物质分类信息统计、宏基因组中样本分类在门纲目科属的各分类层次数目统计(图2-E)以及多个样本的蛋白质功能注释树形比较图等(图2-F)。
3.2 EBI Metagenomics
EBI Metagenomics[15]是欧洲生物信息学研究所(EBI)搭建的分析和存储宏基因组数据的分析平台。用户经注册后可以提交自己产生的宏基因组数据,提交后系统将自动存档数据到欧洲核酸存档 库 中(European Nucleotide Archive,ENA), 并将自动分配数据登录号以便于数据公开。提交到EBI Metagenomics的数据最后都必须要公开,但是用户可以选择一个不长于2年的数据保密期。EBI Metagenomics提供的分析宏基因组数据流程主要包括以下几步:(1)数据质量控制,如去除或者截断低质量的序列,序列片段长度过滤等;(2)通过rRNASelector程序对测序的宏基因组序列片段进行核糖体RNA(rRNA)筛选,然后针对rRNA和非rRNA序列分开处理;(3)针对rRNA序列,使用QIIME软件包对其中的16S rRNA序列进行分类分析,获得宏基因组样本中包含的物种类别;(4)对于非rRNA序列,使用FragGeneScan软件预测其蛋白质编码区域,并使用InterProScan程序预测这些蛋白质的功能结构域和进行功能分析。除了单个样本数据的分析,EBI Metagenomics还提供一个比较分析工具,可以选择已经存储在该数据库中的某个项目中的多个样本数据进行比较分析。这个比较分析主要是对宏基因组数据中蛋白编码序列的Gene Ontotolgy注释进行比较分析。截至2015年9月,EBI Metagenomics中存储的可以公开访问的数据有来自127个项目的4 514个样本的数据,样本来源于土壤、海洋等环境微生物和人的肠道微生物等。对于每个已经公开的数据,其基本的分析结果也可以浏览和下载,包括数据质量控制结果、多种图形方式展示的物种分类结果和功能分析结果等。
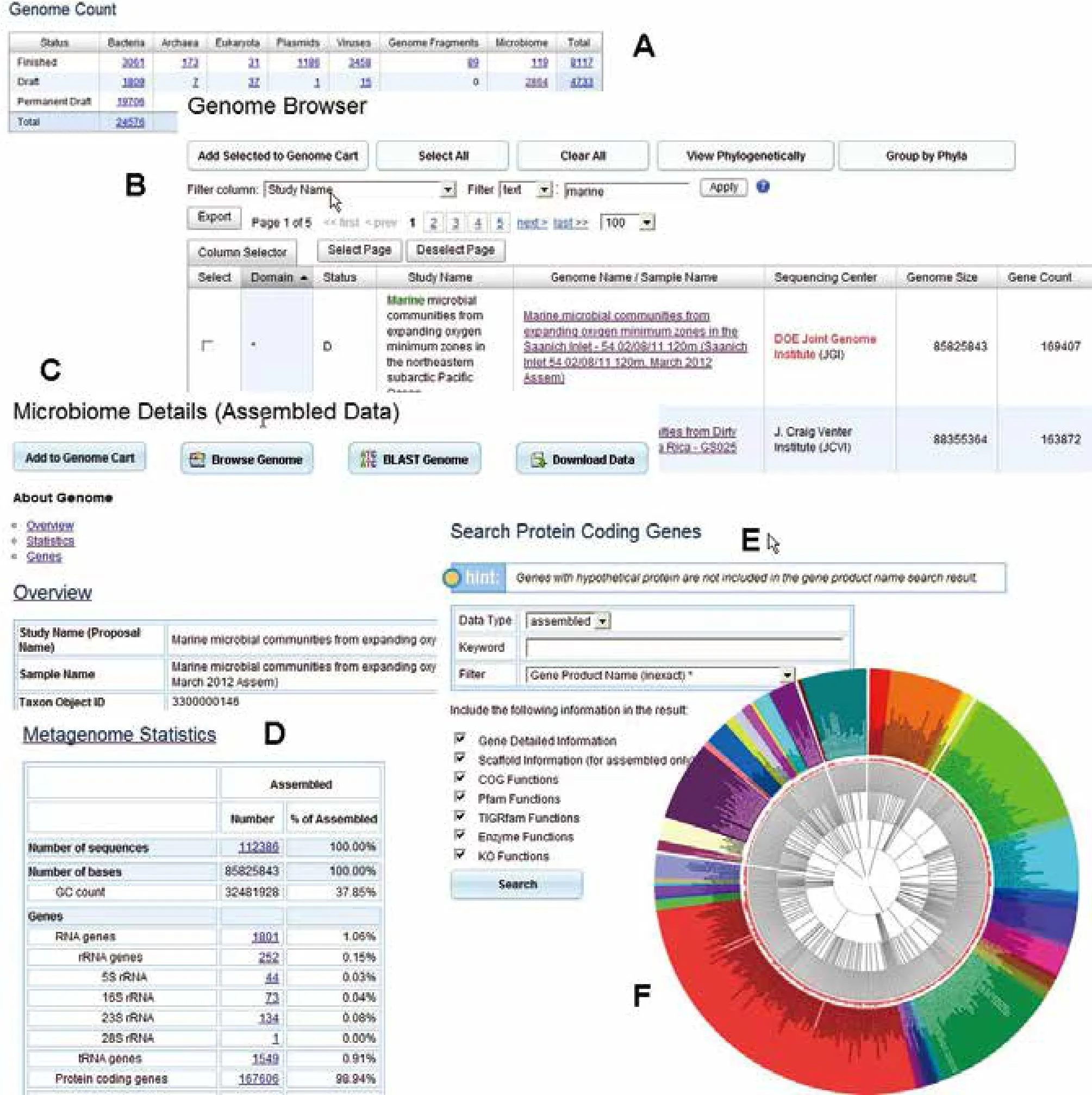
图1 IMG系统中的宏基因组数据详细信息浏览
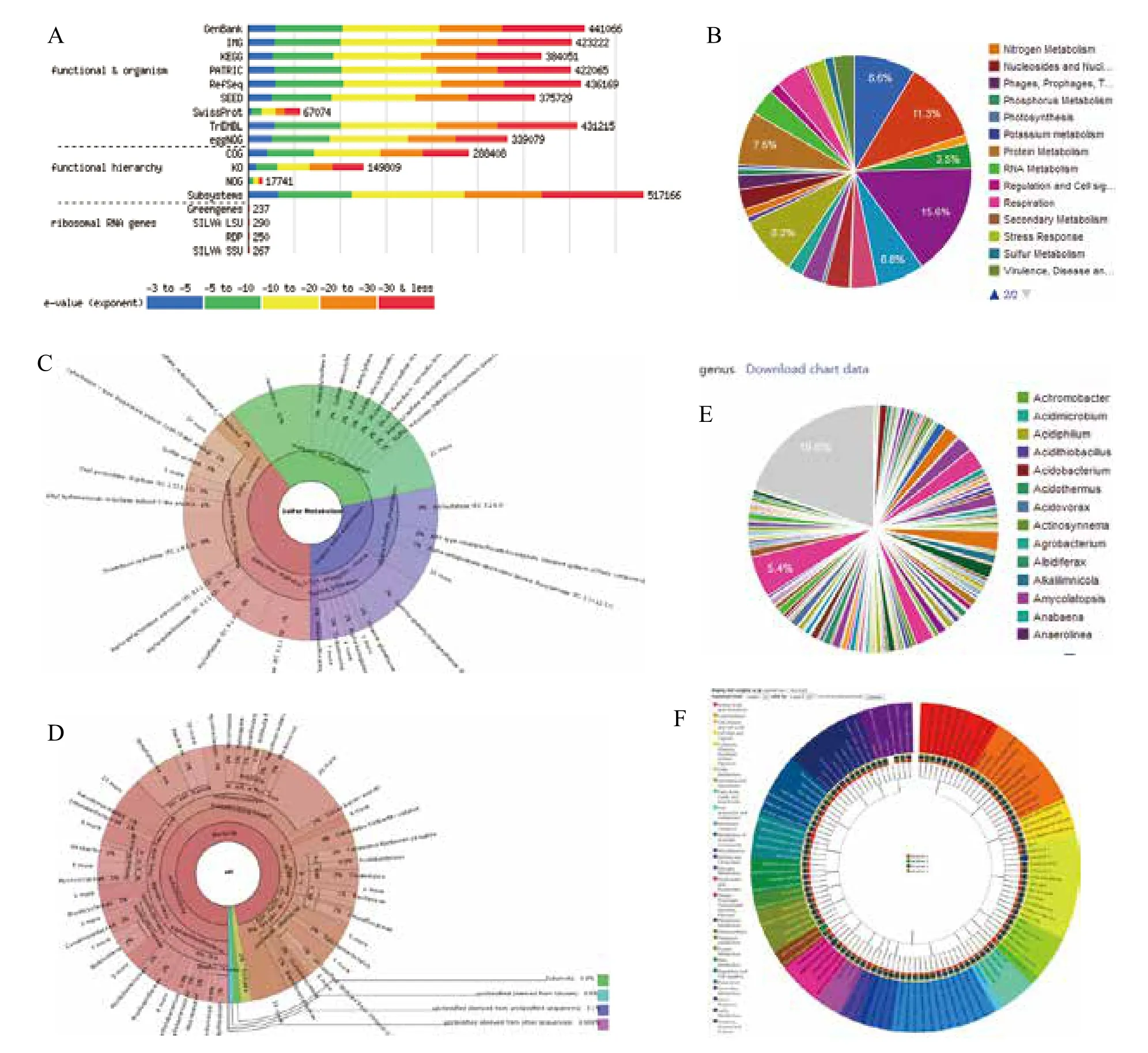
图2 MG-RAST宏基因组注释系统的分析结果
3.3 宏基因组分析仪数据库(MeganDB)
宏基因组分析仪数据库(The MEtaGenome Analyzer Data-Base,MeganDB)是一个为宏基因组分析工具MEGAN[16]特别设计的宏基因组数据库。MEGAN是一个分析宏基因组分类学和功能的独立下载软件包,目前已经更新到第5版本,使用也比较广泛。MEGAN使用NCBI的分类系统进行物种分类,同时使用SEED、KEGG、COG等多个系统进行功能注释,以及进行其他一些比较、聚类和画图等分析。MeganDB数据库目前存储了235套宏基因组数据,并提供了一个Java网络版本的MEGAN程序,可以对一个或多个选择的宏基因组样本数据进行分析,也可以使用MEGAN分析用户自己的数据(<100 MB)或比较公共数据与用户自己的样本数据。MEGAN服务器可以用于上传数据和下载已有宏基因组数据,也可以查询和启动分析这些数据。
3.4 CoMet
CoMet[17]是一个快速进行宏基因组功能谱比较分析的在线分析平台。比较方便的是使用CoMet平台不需要注册即可上传宏基因组数据分析。CoMet平台对于用户上传的FASTA格式的DNA序列进行基因预测,然后预测其中编码的Pfam功能结构域,最后再进行统计分析和比较。值得注意的是CoMet没有上述数据库的保存数据功能,用户提交用于分析的数据在2个月后将自动删除,以节省空间。
4 重点数据库的比较分析
上述内容分别介绍了微生物宏基因组数据库和平台,接下来对其中几个较大的数据库进行比较分析。全球海洋采样项目GOS的目标是评估海洋微生物群落的遗传多样性,其大量的原始数据存储于NCBI SRA数据库中,同时也存储于http://data.imicrobe.us/,具体数据见http://mirrors. iplantcollaborative.org/browse/iplant/home/shared/ imicrobe/projects/26/CAM_PROJ_GOS.read.fa。但是这两个网站都只能下载原始数据,没有分析的结果,用户只能自己根据需要下载原始测序数据分析。海洋生命普查CoML项目的普查对象也是海洋,但是不只限于微生物,还包括其他生物。其目的是评估和解释全世界海洋生命的多样性、分布和丰富性。样本来自多个海域和不同地理环境,如北冰洋、深海、大陆边缘、珊瑚礁等。其中的ICoMM项目是国际海洋微生物普查项目,旨在促进一个能加速发现、理解和意识到海洋微生物的全球意义的议程和环境[18]。ICoMM项目数据存储在https://vamps.mbl. edu/portals/icomm/icomm.php/microbis/网 站 中, 需要注册才能使用。IcoMM的信息主要包含 VAMPS(Visualization and Analysis of Microbial Population Structures)这一整合的数据库和工具集,其提供了微生物的序列以及工具用于分析和可视化微生物群落结构,主要包括可视化分析和数据匝道(data ramp)2个必要元件。可视化分析包括基于对单个微生物群落的物种分类或独立起源的可操作物种单元(Operational taxonomic units,OTUs)所做的分析热图、饼图、多样性估计、稀释曲线和表格数据输出等。数据匝道是研究者将其自己的数据(序列或者物种分类数据)导入VAMPS网站使之与目前共享的数据合并用于单独或者比较分析。另一个大的项目地球微生物组计划EMP,其收集和测序的样本是来自不同地球环境和生态系统的微生物,然后同时使用宏基因组、宏转录组和扩增子测序分析,产生了全球基因图谱描述每个生物群落的蛋白质和环境代谢模型,以及重建的微生物基因组。所以EMP项目的数据类型更多,除宏基因组外,还有宏转录组、代谢组、蛋白质和代谢模型等。
IMG Data Management 和MG-RAST作为两个存储较多微生物数据并且提供分析的平台,它们都能分析数据库本身存储的数据,也可以分析用户上传的数据。但是分析内容有些不同,IMG分析内容比较多,包括整合KEGG、PFAM等多个功能资源对数据进行鉴定和注释,同时也有基本的数据统计如序列碱基数目、G+C含量等,还可以进行基因组组装并展示基因组在系统发生学上的分布。而MG-RAST则主要是分析宏基因组数据中的物种分布类别组成,但是它提供了树图、柱状图、热图、组成分分析图和表格等方式展示结果,结果形式多样化。
5 展望
宏基因组学是研究环境微生物的一个重要手段,这些宏基因组数据的解析依赖于很好的分析工具。目前虽然有不少宏基因组分析工具,但还是存在一些不足。对宏基因组测序数据处理最理想的结果就是通过组装软件组装出其中各种微生物的基因组。但是目前还远远做不到,只能在很大测序数据量的情况下对少数序列差异较大的物种组装有较好效果。组装的效果一方面依赖于测序的深度和序列片段长度的加大,另一方面依赖于组装算法的改进,需要能在宏基因组这样的混合样本中辨别出单个基因组序列并组装。基因组组装后需要进行基因预测,目前也有很多基因测序软件,但是宏基因组中不同物种可能有不同的密码子偏好性和物种特异的基因,因此需要有适应性更广的预测算法或者同时使用多种预测软件和训练数据进行预测。宏基因组数据中的物种分类也是其中一个关键分析,目前有基于基因、参考基因组比较、序列组成等多种方式的物种分类[19]。基于宏基因组数据中预测到的基因的相似性注释分类是目前最为普遍的物种分类方法。
上述宏基因组数据分析工具都需要有一定的生物信息学基础和技能才能分析,这也是目前困扰环境微生物研究者的问题之一。因此,将已有的环境宏基因组数据建立方便易用的在线公开数据库,搭建操作简单的网络版本宏基因组学分析平台是非常有必要的,也是研究共享的必然趋势。可喜的是,国际上已有几十个相关数据库和平台,这些资源存储了许多重要的国际合作项目或者个人研究项目的环境宏基因组学数据,其总的数据量达到上百万个样本的级别,样本类型也是多种多样,包括海洋、陆地、极端环境、油田等环境的样本。此外,除了上述介绍的专门存储环境微生物宏基因组数据的数据库,NCBI的SRA数据库也专门存储各种高通量测序的数据,包括各种基因组和转录组数据,其中含有约6 000组非肠道微生物的环境宏基因组数据集。另一方面,这些数据的采样和产生都耗费了大量人力物力和经费,充分利用它们也是对资源的重利用,可以减少消耗,提高效率,节省各方面的开销和能源。
如何充分利用和挖掘这些数据,对它们进行综合的二次深度分析,获得新的发现,是一个值得思考的问题。目前环境宏基因组领域内的一个比较大的问题是数据分散在不同的数据库中或者分布在不同的研究者手中。如果能收集所有数据并从不同的角度整理这些数据,如从环境类型、数据类型等角度整合这些数据,将能更加方便的为其他研究者提供便利。例如,研究热泉环境的微生物就可以直接提取热泉环境的宏基因组数据集进行整合分析,而不需要花费大量精力收集数据,甚至重新采集样本测序。合理有效的整合这些不同来源和类型的环境宏基因组数据将是一个发展方向和趋势。本文介绍的这些环境宏基因组数据库就是这方面的尝试,介绍和了解这些不同大项目的数据或者不同研究者来源的数据。用户可以根据自己的需求综合这些数据库进行使用,获得最全面的所需要的数据。利用已有的宏基因组学数据从不同的角度整合、比较和分析发现,新的微生物资源及其可利用规律,必将推动环境微生物的研究。
[1] 孙欣, 高莹, 杨云锋. 环境微生物的宏基因组学研究新进展[J].生物多样性, 2013, 21(4):393-400.
[2] Handelsman J, Rondon MR, Brady SF, et al. Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J]. Chemistry & Biology, 1998, 5(10):R245-R249.
[3] Chistoserdova L. Recent progress and new challenges in metagenomics for biotechnology[J]. Biotechnology Letters, 2010, 32(10):1351-1359.
[4] Gilbert JA, Jansson JK, Knight R. The Earth Microbiome project:successes and aspirations[J]. BMC Biology, 2014, 12(1):69.
[5]Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea[J]. Science,2004, 304(5667):66-74.
[6]CoML. First census of marine life 2010:highlights of a decade of discovery[R]. Washington:Census of Marine Life, 2010.
[7]Reddy TBK, Thomas AD, Stamatis D, et al. The Genomes OnLine Database(GOLD)v. 5:a metadata management system based on a four level(meta)genome project classification[J]. Nucleic Acids Research, 2015, 43 (D1): D1099-D1106.
[8]Sun S, Chen J, Li W, et al. Community cyberinfrastructure for advanced microbial ecology research and analysis:the CAMERA resource[J]. Nucleic Acids Research, 2011, 39(suppl. 1):D546-D551.
[9]Parthasarathy H, Hill E, MacCallum C. Global ocean sampling collection[J]. PLoS Biology, 2007, 5(3):e83.
[10]Wommack KE, Bhavsar J, Polson SW, et al. VIROME:a standard operating procedure for analysis of viral metagenome sequences[J]. Standards in Genomic Sciences, 2012, 6(3):427.
[11]An D, Caffrey SM, Soh J, et al. Metagenomics of hydrocarbon resource environments indicates aerobic taxa and genes to be unexpectedly common[J]. Environmental Science & Technology,2013, 47(18):10708-10717.
[12]Markowitz VM, Chen IMA, Chu K, et al. IMG/M 4 version of the integrated metagenome comparative analysis system[J]. Nucleic Acids Research, 2014, 42(D1):D568-D573.
[13]Meyer F, Paarmann D, D’Souza M, et al. The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes[J]. BMC Bioinformatics,2008, 9(1):386.
[14]Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the Project to Annotate 1000 Genomes[J]. Nucleic Acids Research, 2007, 33(17):5691-5702.
[15] Hunter S, Corbett M, Denise H, et al. EBI metagenomics--a new resource for the analysis and archiving of metagenomic data[J]. Nucleic Acids Research, 2014, 42(D1):D600-D606.
[16]Huson DH, Auch AF, Qi J, et al. MEGAN analysis of metagenomic data[J]. Genome Research, 2007, 17(3):377-386.
[17]Lingner T, Asshauer KP, Schreiber F, et al. CoMet--a web server for comparative functional profiling of metagenomes[J]. Nucleic Acids Research, 2011, 39(8):W518-523.
[18]Amaral-Zettler L, Artigas LF, Baross J, et al. A global census of marine microbes[M]// Life in the World’s Oceans:Diversity,Distribution, and Abundance. Wiley-Blackwell, 2010.
[19]Teeling H, Glöckner FO. Current opportunities and challenges in microbial metagenome analysis--a bioinformatic perspective[J]. Briefings in Bioinformatics, 2012, 13(6):728-742.
(责任编辑 马鑫)
An Introduction to Metagenome Databases of Environmental Microbiology
Wang Huili1Guo Anyuan2
(1. Department of Environmental Engineering,Urban Construction Engineering Division,Wenhua College,Wuhan 430074;2. College of Life Science and Technology,Huazhong University of Science and Technology,Wuhan 430074)
The huge data produced by metagenome are valuable resources for environmental microbiology research. Until now, there are many big projects such as the Earth Microbiome Project and Census of Marine Life, which generated huge metagenome data and also constructed various databases and platforms to store and analyze these data. In this review, we summarized the current big projects, databases and online analysis platforms for environmental metagenomes. We introduced the project background, the sample information, the data type, the usage mode and the webpage of those databases.
metagenome;database;resources;microbiology
10.13560/j.cnki.biotech.bull.1985.2015.11.008
2015-08-27
科技部科技基础性工作专项(2012FY112900)
王慧丽,女,硕士,研究方向:环境微生物和水处理;E-mail:lypp1229@126.com
郭安源,男,博士,研究方向:生物信息和数据库;E-mail:guoay@hust.edu.cn
