孕烷X受体对细胞色素P450 CYP3A基因表达的调控在中药配伍禁忌及中药毒性早期预测中的应用
2015-08-31王宇光天津中医药大学天津330007军事医学科学院放射医学研究所北京100850
李 晗,王宇光,高 月(1.天津中医药大学,天津 330007;.军事医学科学院放射医学研究所,北京 100850)
孕烷X受体对细胞色素P450 CYP3A基因表达的调控在中药配伍禁忌及中药毒性早期预测中的应用
李 晗1,2,王宇光2,高 月2
(1.天津中医药大学,天津 330007;2.军事医学科学院放射医学研究所,北京 100850)
中药配伍禁忌是药物与机体相互作用本质的具体体现,是中药配伍理论的重要组成内容。毒性早期预测是药物安全性评价的重要组成部分。快速、早期获得药物可能的毒性反应数据,建立一种简单、可靠地中药配伍禁忌和中药毒性早期预测方法,是当今毒理学领域研究的重点之一。孕烷X受体(PXR)是一种配体依赖性转录因子,大量临床药物作为其配体或激活剂通过激活PXR而诱导细胞色素P450 CYP3A (CYP3A)基因表达。联合用药过程中,PXR的激活可能会增加药物发生相互作用和不良反应的风险,导致药效降低甚至产生毒性。本文从PXR-CYP3A途径为药物相互作用研究提供分子学机制,以及为中药配伍禁忌的研究和药物毒性早期预测提供了新思路。
中草药;药物配伍禁忌;毒性早期预测;细胞色素P450 CYP3A;孕烷X受体;药物代谢
随着中药的使用日益广泛,中药安全性问题与中药不良反应逐渐被人们所关注。中药含有丰富的天然活性成分,具有多种生理功能,但是中药成分和药理作用复杂,长期使用或与化学药物联用时可能对药物代谢酶的表达或活性产生影响,降低药效甚至产生毒性。传统的用于中药配伍禁忌研究以及中药早期毒性预测的常规毒理学实验方法因灵敏度低、周期长、花费多且需消耗大量实验动物等缺陷而难以满足现代药物开发中进行高通量筛选的需求。核受体能结合经药物设计而被修饰的小分子,从而调控相关疾病如糖尿病、代谢性疾病、骨质疏松、癌症等[1],并参与内源性物质和多种外源性物质,如药物、污染物、致癌物质、杀虫剂或环境化学药品的代谢[2]。其中核受体家族中孕烷X受体(pregnane X receptor,PXR)作为关键的转录调控因子主要参与药物代谢酶基因表达,改变酶活性,进而降低药效甚至产生毒性。因此,有关基于PXR介导药物代谢酶调控基因表达机制的研究成为了热点,特别其对细胞色素P450 CYP3A(cytochrome P-450 CYP3A,CYP3A)在药物的减毒、增毒与早期毒性预测方面的作用,更是毒理学和分子药理学研究的重点。
1 中药配伍禁忌
一段时间以来,由于公众对中药毒性知识匮乏,对中药配伍禁忌更是知之甚微,在临床治疗过程中也对其安全性缺乏有效的监控。近些年,随着国内外中药“药害”事件的不断报道,国内外公众对中药安全性问题开始引起关注。中药配伍禁忌的科学性是现代药性理论争论的焦点之一,然而现阶段对其研究主要停留在文献挖掘、物质基础、生物效应以及毒性考察的阶段,尚未对其内在机制做深层次研究。目前针对中药配伍禁忌的方法与思路主要采用药物代谢动力学方法。作为经典的药物代谢动力学研究方法之一,中药有效成分血药浓度法适用于已明确有效成分或毒性成分的配伍中药,例如黄连解毒散[3-4]和银杏叶[5]。该法主要包括高效液相色谱法、液-质联用法、气-质联用法、分光光度法、薄层扫描法、毛细管电色谱法以及原子吸收光谱法等。但因为仪器分析法检测出的化学成分只是中药配伍复方中繁多化学成分的一种或几种,而不能全面反映中药配伍所导致的有效成分或毒性成分的变化情况,不能百分百代表复方药物代谢动力学,并且不能检测有效成分不明的中药配伍的药物代谢。生物效应法也是研究中药配伍禁忌的主要方法。中药复方的组成成分主要分为有效成分和毒性成分,通过检测2类成分的生物活性可以推测出复方整体在体内的大致过程。基于此原理,生物效应法主要分为药理效应法和毒理效应法。药理效应法是通过判断药物的药效强度,以时间-效应关系和剂量-效应关系转换为时间-剂量关系,从而得到药物代谢动力学参数的方法。该法可将中药在体内的动态变化药理指标可逆且定量地反映出来,有利于有效成分尚不明确的中药复方研究[6-7]。毒理效应法则是将动物急性死亡率和血药浓度法中多点测定原理相结合,测定药物蓄积性的方法,并以急性死亡率为判断指标,即通过设计多组动物不同时间间隔的给药方法,得出体内药物的百分率在不同时间的动态变化,由此推算出药动学参数。该药物动力学研究方法适用于有效成分尚未明确以及成分繁多复杂的中药复方研究,尤其适用于无合适化学定量方法的毒性中药及复方的研究,但因需要大量动物实验,工作量繁重,并不能满足快速评价复方的需求[8-9]。所以,建立一种基于体外培养的快速筛选评价平台对于中药配伍禁忌研究具有重要意义。
2 中药毒性早期预测
药物研发是一个高投入、高回报、高难度的产业。在药物临床前阶段,毒性问题是决定研发成败的关键。所以,在研发阶段尽早发现化合物的潜在毒性已成为毒理学研究的热点。动物实验是传统药物毒性预测常用的方法之一,但是采用实验动物病理学及其相关生化指标来判断药物的毒性,存在工作量繁重、耗时长、评价效率低等情况。更重要的是,因为种属差异,从动物实验中获得的数据并不能对人体发生的实际情况进行全面准确地预测[10-11]。20世纪90年代开始,随着现代医学技术的发展,在新药发现阶段,国外制药公司采用毒理学与药动学、药效学、药理学相结合的方法对新化学实体(new chemical entities,NCE)进行筛选和优化。通过综合毒理学、药动学、药效学及药理学的各项结果分析NCE的开发前景,挑选出候选新药进行结构优化改造和进一步的后续研究。研究主要采取的方法有高通量与高内涵毒性筛选技术方法,例如细胞毒性筛选技术、一般毒性筛选技术、特殊毒性筛选技术等;现代组学技术方法,主要包括基因组学、蛋白质组学和代谢物组学技术,可分别预测药物在基因水平、蛋白质水平和机体代谢这3个水平的毒性并对毒性作用机制进行深入研究;评价药物的慢性毒性、致癌性和毒性作用机制的“基因敲除”和转基因动物模型方法;以及毒性的定量结构活性模型(quantitative structure activity relationships,QSAR)和计算机辅助毒理学(in silico toxicology)的方法。与化学药在毒性预测与评价研究领域的突飞猛进相比,中药在此领域刚刚起步。随着中药使用的日益广泛,临床出现毒性甚至引起死亡等报道不断增多,中药安全性问题逐渐被人们所关注。传统观念认为,相比化学药物,中药更为安全,不良反应更少[12]。但是中药也是由各种化学成分所组成的,这些化学成分在体内发挥治疗作用的同时,也可能会产生不良反应,甚至损害某些组织和器官。因此,急需一套快速的中药毒性预测方法。
3 CYP3A与其转录调节的关键因子PXR
3.1 CYP3A
人体特别是肝含有多种氧化酶和结合酶,其中CYP-450是许多同工酶组成的超家族,其对药物代谢和药物之间相互作用有着重要影响。作为外源性化合物的主要代谢酶,CYP酶主要存在于肝。在CYP超家族中,CYP3A亚型与药物代谢最相关。CYP3A是细胞色素CYP超家族的主要成员,在内源性激素、胆汁酸和外源性化学物以及临床药物的代谢中起着关键作用[13-14]。CYP3A占肝微粒体P450总量的25%~28%,参与≥50%临床常用药物的代谢,目前已明确约有150多种药物通过CYP 3A代谢,主要包括钙拮抗剂、免疫抑制剂、大环内酯类抗生素、甾体避孕药和抗病毒药等。CYP3A可在保护机体在生理性高激素水平条件下免受损害,促进有毒化学物的降解等方面起到重要的作用[15]。另一方面,许多结构差异较大的化学物可诱导或抑制CYP3A的活性,这在临床上可引起药物间相互作用,降低药效甚至无效,严重者则产生毒性作用[16-17]。
3.2 PXR
核受体是一类能与DNA应答元件结合的配体激活性转录调控因子,它们的主要功能是特异性调控发育、生殖、代谢相关基因的表达。其中包括许多重要的药物靶分子,如维生素D受体、糖皮质激素受体和雌激素受体等[18]。PXR属于核受体超家族(nuclear receptor superfamily,NR)中NR1Ⅰ亚家族的成员之一,参与大量的内源性和外源性化学物质的生物转化。1997年,Kliewer等[19]克隆出小鼠PXR蛋白的全长互补DNA(cDNA),鉴于此受体可被一系列天然化合物或合成的孕烷所激活,故将其命名为PXR。几年后,大鼠、家兔和犬等不同种属动物及人的PXR也被相继克隆。PXR是由配体结合域(ligand binding domain,LBD)和DNA结合域(DNA binding domain,DBD)组成。当外源物进入细胞后,直接与核内LBD结合,形成受体-配体复合物,然后再结合视黄醛X受体(retinoid X receptor,RXR)形成异二聚体,最后该二聚体与CYP3A基因启动子上的PXR反应元件(PXR response element,PXRE)结合,诱导CYP3A基因的表达(图1)。
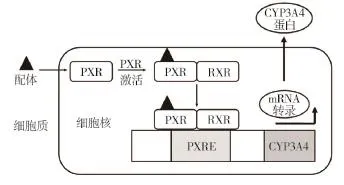
图1 孕烷X受体(PXR)激活靶基因细胞色素P-450 CYP3A(CYP3A)过程。RXR:视黄醛X受体;PXRE:PXR反应元件
与其他核受体不同,PXR的LBD结构较为特殊,两股β折叠将伸直的螺旋插入一个可变的环中从而形成球状配体结合腔,此腔具有潜在的扩展性,因此可容纳各种大小的配体,具有潜在的扩展性,同时又与PXR配体的广泛性有关[20]。研究显示,哺乳动物PXR的DBD具有高度保守性,大约96%氨基酸序列相同,而LBD则存在明显的种属差异。人和大鼠PXR的LBD仅有76%氨基酸序列相同,提示不同种属的PXR虽可调控相同或类似的靶基因表达,但配体的结合活性则具有较大的差别。另外研究发现,特定的氨基酸序列是决定LBD中特异性配体结合效能的首要因素。
3.3 PXR对CYP3A转录调控
研究显示,PXR可通过调节CYP3A的转录活化保护机体免受潜在有毒化学物的危害。一系列证据表明PXR为CYP3A基因关键的调控因子。首先,选择性地表达PXR的组织和表达丰度与CYP3A完全一致,主要分布于哺乳动物的肝、小肠和结肠组织中[21]。其次,PXR能与CYP3A基因启动子上的PXR反应元件结合[22]。研究发现,PXR结合于啮齿类动物CYP3A基因序列上的启动子和(或)增强子上调的直接重复序列3(direct repeat 3,DR3)或结合于人的倒向重复序列6(everted repeat 6,ER6)。一旦PXR与这些部位结合,则会诱导CYP3A的转录。第三,多数CYP3A诱导剂均能结合并激活PXR,如利福平、苯巴比妥、克霉唑和磺吡酮(苯磺唑酮,sulfinpyrazone)等。最后,在体实验研究发现,敲除自身pxr基因小鼠对鼠或人特异CYP3A的诱导剂均无响应,cyp3a mRNA的基础水平没有增加,但是转染了人PXR(hPXR)后,人特异诱导剂对CYP3A产生诱导作用[23]。综合以上结果可知,核受体家族成员PXR可能在CYP3A转录激活机制中发挥核心作用。
4 PXR-CYP3A途径在中药配伍禁忌及中药毒性早期预测中的应用
中药配伍对药物代谢酶的影响可导致药效的降低甚至产生毒性,所以从药物代谢酶及其相关核受体的调控角度对配伍降效、减效和增毒的机制进行研究是揭示中药配伍禁忌以及中药毒性的重要环节。鉴于CYP3A4参与60%药物的代谢,是体内最重要的药物代谢酶之一,PXR介导的CYP3A调控途径被认为是临床药物相互作用的重要分子基础。同时PXR可被多种中药激活,调控下游靶基因CYP450的表达,使药物疗效降低或消失,从而导致不利的药物相互作用的产生,因此,PXR在中药配伍禁忌与中药毒性早期预测方面具有重要作用[24]。该方法简单,操作方便,反应时间短,易于大量实验及多次重复,能直接反映药物对P450酶的诱导或抑制作用,并且它适合于所有通过CYP450酶代谢的药物的研究。研究者不仅要在临床用药阶段对PXR与药物相互作用关系进行研究,更应在新药研发与毒性早期预测阶段对化合物与PXR-CYP3A的相互作用进行深入研究。当前,已有多种针对PXR-CYP3A途径的研究技术体系被相继建立并广泛应用。
寻找药物潜在的代谢物和药物生物转化途径的研究中,以及新药在临床广泛使用前,通常采用原代培养人肝细胞检测这种药物对CYP3A4基因表达是否具有诱导作用,原代培养人肝细胞是一种简单而有效的模型。将原代肝细胞和药物孵育,能得到相对大量高纯度的代谢物,此法减少外界因素的干扰。研究显示,当归的有效成分白当归素(byakangelicn)能显著上调人原代肝细胞中的CYP3A4 mRNA及蛋白表达水平[25]。但人源肝细胞不易获得,培养繁琐且耗时,很难避免不同肝组织捐献者之间肝细胞质量的个体差异。动物原代肝细胞又不能完全反映人CYP3A4的诱导特性。研究者将构建成功的碱性磷酸酶、半乳糖苷酶或荧光酶的报告质粒与含PXR序列表达质粒共转染于细胞中,在化合物诱导下,根据报告基因中特异性酶的表达情况,间接测定化合物诱导CYP3A的能力。此法可对具有PXR激活作用的先导化合物进行初步筛选,从而快速、高效缩小下游的研究范围。贯吐连翘(圣约翰草,Hypericum perfuoretum,St.John′s wort),主要用于治疗抑郁和炎症的非处方药。报告基因分析显示,其能激活PXR并诱导CYP 3A4的表达。联合用药时,可增加其他合用药物的代谢率和清除率,例如,口服避孕药、抗艾滋病药物利托纳韦和英地纲韦及免疫抑制剂环孢素[26]。但由于每次都要进行质粒转染,操作繁琐,又不能保证稳定性和重复性。而且,许多其他因子也对CYP3A转录活化产生影响。
研究者又建立基于PXR-CYP3A4通路的工程细胞株模型的稳定转染的报告基因方法。利用Gaussia分泌型荧光素酶报告基因pGL4.17载体,将人PXR表达载体和报告基因载体共转染HepG2细胞,通过G418压力筛选获得稳定转染细胞株后进行单克隆培养。通过利福平和地塞米松等PXR激动剂验证实验,以及优化培养条件,建立药物相互作用体外快速筛选模型。此模型可在药物研发早期对化合物进行快速、大量的PXR活性筛选,间接判断化合物能否诱导CYP3A。研究者采用此法检测了118种中药化学成分标准品后发现,靛蓝、百秋李醇、胡椒碱、千金藤素、五味子酯甲、白花前胡甲素、菊苣酸梣酮和银杏酸的PXR激活效应接近利福平[27]。由于报告基因检测技术是基于转录水平的筛选方法,其结果并不能完全代表翻译水平以及酶活性水平的最终结果,因此具有一定的局限性[28]。
离体实验只能部分反映临床现象,其结果无法完全替代动物实验。离体培养人肝细胞过程中,各种生理特性也会发生改变,包括药物转运体表达下降,肝药酶活力降低等,使其结果往往不能很准确反映药物代谢和毒性现象[29-30]。近几年,应用基因敲除和转基因技术建立了人源化转基因小鼠模型,此模型可使小鼠模拟出人的药物代谢反应性,因而被认为是体内判断化合物是否能诱导CYP3A的理想模型。此模型的建立,可减少体外实验造成的误差以及种属差异,使研究更便捷,值得进一步推广[31]。
应用这些评价PXR活性的方法可为预测某种新药与其他药物联合使用时是否会产生相互作用提供线索,为药物毒性早期预测提供新的思路。
5 结语
虽然上述方法还存在一些不足,需要进一步改善。但是随着医学科技的不断发展,研究者可对PXR与药物代谢酶基因的调控作用进行更深入的、系统的研究,在分子水平上明确PXR减少外源性化合物在体内蓄积的原因,进一步阐明药物相互作用的机制,建立更多基于PXR核受体在中药配伍禁忌以及中药早期毒性预测中的方法,及早在药物研发阶段获得相关药物的毒性数据,缩短研究周期,减少药物研发成本,并降低临床使用过程中的不良反应,进而提高中药使用的安全性。
[1]Zhang Y,Qin Q,Zhou DJ,Chen B,Chen M. Progress in nuclear receptor superfamily and nuclear receptor regulator[J].J Regional Anat Operative Surg(局解手术学杂志),2007,16(1):58-59.
[2]Waxman DJ,Azaroff L.Phenobarbital induction ofcytochromeP-450geneexpression[J]. Biochem J,1992,281(Pt3):577-592.
[3]Lin XL,Ma YF,Yu DJ,Yao JS,Huang XH,Huang YF.Pharmacokinetics study on geniposide as the effective ingredient of ultramicro pulverised powder of Huanglian Jiedusan in rabbits in vitro[J].Sci Technol Rev(科技导报),2011,29(5):51-55.
[4] Zhang Z,Yan YF,Chen KJ.Effects of Chuanxiong-Chishao dispensing ratio on the pharmacokinetics of Paeoniflorin in the canine[J].China J Chin Mat Med(中国中药杂志),2000,25(11):688-691.
[5] Zhang Y,Liu JX,Lin L,Zhang QQ,Wang JN. HPLC-ECD Determination of flavonols from Ginkgo biloba leaf extracts in rat plasma and its pharmacokinetic study[J].Chin J Pharm Anal(药物分析杂志),2011,31(1):10-14.
[6] Ren P,Huang X,Jiang YP,Guan AD,Song GZ. Effect of Sijunzi Decoction on motilin and pharmacokinetic characteristics of tetramethylpyrazine in rat model of spleen deficiency syndrome[J]. Chin J Integrated Tradit West Med(中国中西医结合杂志),1997,17(1):45-47.
[7]Huang X,Chen KY.Scientific evidence,elements,significance and prospects of the new hypothesis of compound effect composition dynamics[J].China J Chin Mater Med(中国中药杂志),1997,22(4):250-252.
[8] Xu KJ,Sun KX,Lu YC,Zhang HJ,Wu LH,Hu JQ. Study on human bioavailability ofthe injection and the aerosol of″Shuang Huang Lian″[J].Chin J Hosp Pharm(中国医院药学杂志),1992,12 (11):484-486,526.
[9]Wang XF,Qin J,Yang CM.Bacterioassay of pharmacokinetic parameters of methyl hydroquinone in rabbits[J].Northwest Pharm J(西北药学杂志),1997,12(2):70-71.
[10]Matheis KA,Com E,Gautier JC,Guerreiro N,Brandenburg A,Gmuender H.Cross-study and cross-omicscomparisons of three nephrotoxic compounds reveal mechanistic insights and new candidate biomarkers[J].Toxicol Appl Pharmacol,2011,252(2):112-122.
[11] Kong QX,Yao QS,Huang YY,Li RR,Li M,Lv J,et al.Request for peer review on toxicological pathology[J].Drug Eval Res(药物评价研究),2011,34(5):370-373.
[12] Wang YG,Ma ZC,Liang QD,Liu M,Liu BB,Tan HL.Study on the eighteen Incompatible pairs based on cytochrome P450[J].World Sci Technol Mod Tradit Chin Med Mater Med(世界科学技术-中医药现代化),2011,13(1):36-40.
[13] Wang JS,Xu ZH,Zhou HH.Cytochrome P450 3A4 and drug metabolism[J].Chin J Clin Pharmacol(中国临床药理学杂志),1996,12(4):231-235,241.
[14]Peng H,Cheng ZN.Drug interaction related to CYP3A4[J].Chin J Clin Pharmacol(中国临床药理学杂志)2001,17(5):379-385.
[15] Xie W,Radominska-Pandya A,Shi Y,Simon CM,Nelson MC,Ong ES.An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids[J].Proc Natl Acad Sci USA,2001,98 (6):3375-3380.
[16] Dresser GK,Spence JD,Bailey DG.Pharmacokineticpharmacodynamicconsequencesandclinical relevance of cytochrome P450 3A4 inhibition[J]. Clin Pharmacokinet,2000,38(1):41-57.
[17] Dai D,Tang J,Rose R,Hodgson E,Bienstock RJ,Mohrenweiser HW,et al.Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos[J].J Pharmacol Exp Ther,2001,299(3):825-831.
[18] Willson TM,Kliewer SA.PXR,CAR and drug metabolism[J].Nat Rev Drug Discov,2002,1 (4):259-266.
[19] Kliewer SA,Moore JT,Wade L,Staudinger JL,Watson MA,Jones SA,et al.An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway[J].Cell,1998,92 (1):73-82.
[20] Orans J,Teotico DG,Redinbo MR.The nuclear xenobiotic receptor pregnane X receptor:recent insights and new challenges[J].Mol Endocrinol,2005,19(12):2891-2900.
[21]Wang YG,Wang SQ,Gao Y.The induction of CYP3A regulated by pregnane X receptor and its significance in drug metabolism[J].Acta Pharm Sin(药学学报),2006,41(1):1-6.
[22]Quattrochi LC,Guzelian PS.Cyp3A regulation: from pharmacology to nuclear receptors[J]. Drug Metab Dispos,2001,29(5):615-622.
[23] Xie W,Barwick JL,Downes M,Blumberg B,Simon CM,Nelson MC.Humanized xenobiotic response in mice expressing nuclear receptor SXR[J].Nature,2000,406(6794):435-439.
[24]Zhou SF.Drugs behave as substrates,inhibitors and inducers of human cytochrome P450 3A4 [J].Curr Drug Metab,2008,9(4):310-322.
[25] Yang J,Luan X,Gui H,Yan P,Yang D,Song X. ByakangelicininducescytochromeP4503A4 expressionviatransactivationofpregnaneX receptors in human hepatocytes[J].Br J Pharmacol,2011,162(2):441-451.
[26] Xie HG,Kim RB.St John's wort-associated drug interactions:short-term inhibition and long-term induction?[J].Clin Pharmacol Ther,2005,78 (1):19-24.
[27]Wang YG,Lu BB,Gao Y.Screening of natural products based on receptor and related pharmacological and toxicological studies[C]//The Scientific and Technical Forum of the Fourth China Youth Scholars(中国毒理学会第四届中青年学者科技论坛论文集).Beijing:The Society of Toxicology,2014:43.
[28] Moore JT,Kliewer SA.Use of the nuclear receptorPXR to predict drug interactions[J].Toxicology,2000,153(1/3):1-10.
[29] Richert L,Liguori MJ,Abadie C,Heyd B,Mantion G,Halkic N.Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin,is not affected by hepatocyte cold storage and cryopreservation,but is strongly changed after hepatocyte plating[J].Drug Metab Dispos,2006,34(5):870-879.
[30] Skett P.Problems in using isolated and cultured hepatocytes for xenobiotic metabolism/metabolismbased toxicity testing-solutions?[J].Toxicol In Vitro,1994,8(3):491-504.
[31] Cheung C,Gonzalez FJ.Humanized mouse lines and their application for prediction of human drug metabolism and toxicological risk assessment[J].J Pharmacol Exp Ther,2008,327(2):288-299.
Corresponding authors:WANG Yu-guang,Tel:(010)66932201,E-mail:wyg79@139.com;GAO Yue,Tel:(010)66931312,E-mail:gaoyue@bmi.ac.cn
(本文编辑:乔虹)
Application of pregnane X receptor′s regulation of induction of cytochrome P-450 CYP3A to incompatibility of traditional Chinese medicine and toxicity predetermination
LI Han1,2,WANG Yu-guang2,GAO Yue2
(1.Tianjin University of Traditional Chinese Medicine,Tianjin 330007,China;2.Institute of Radiation Medicine,Academy of Military Medical Sciences,Beijing 100850,China)
The incompatibility of traditional Chinese medicine is an important part of traditional Chinese medicine compatibility theory,while early toxicity predetermination of traditional Chinese medicine is an important component of drug safety evaluation.Once a simple and reliable method to predict the compatibility of Chinese medicine and Chinese traditional medicine is established,it is possible to obtain the data of toxic reaction of Chinese herbal medicine quickly and early.Pregnane X receptor (PXR)is a ligand dependent transcription factor,acting as a ligand or activator of a large number of clinical drugs and of cytochrome P-450 CYP3A(CYP3A)gene expression induced by PXR.In the course of treatment of diseases,the activation of PXR may increase the risk of drug interactions and adverse reactions,resulting in decreased efficacy and even toxicity.This paper may provide a new method of drug toxicity predetermination depending on the PXR-CYP3A pathway.
drugs,Chinese herbal;drug incompatibility;drug toxicity predetermination;cytochrome P-450 CYP3A;pregnane X receptor;drug metabolism
The project supported by National Basic Research Program of China(″973 Program″)(2011CB505304)
R285.1
A
1000-3002-(2015)06-0967-06
10.3867/j.issn.1000-3002.2015.06.014
国家重点基础研究发展计划(973计划)(2011CB505304)
李 晗,女,博士研究生,主要从事新药发现与中药药理学研究,E-mail:wzf38@126.com
王宇光,Tel(010)66932201,E-mail:wyg79@139.com;高月,Tel:(010)66931312,Fax:(010)66931312,E-mail:gaoyue@bmi.ac.cn
(2015-09-02接受日期:2015-11-09)
