氟丙嘧草酯的新合成工艺
2015-08-25
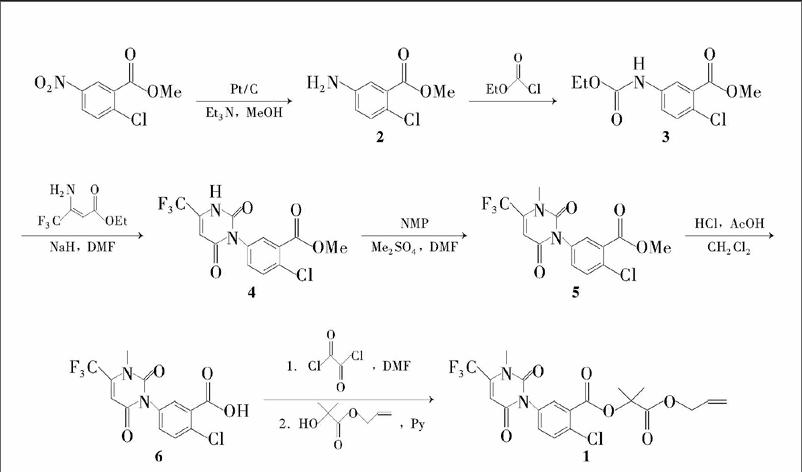
氟丙嘧草酯【{2-氯-5-[1,2,3,6-四氢-3-甲基-2,6-二氧-4-(三氟甲基)嘧啶-1-基]苯甲酸-1-(丙烯氧基羰基)-1-甲基乙基酯}(1)】是一类高效、选择性强以及对环境友好的脲嘧啶类除草剂,属于原卟啉原氧化酶抑制剂,主要用于果园(包括葡萄园)、棉花地和非耕地,芽前处理防除禾本科杂草,以及一年生和多年生阔叶杂草、莎草等。
对于1 的合成,文献方法采用2-氯-5-硝基苯甲酸为原料,经酰氯化、与2-羟基-2-甲基丙酸丙烯酯缩合得2-氯-5-硝基苯甲酸-1-烯丙氧羰基-1-甲基-乙基酯,再经还原、异氰酸化、与3-甲氨基-4,4,4-三氟巴豆酸乙酯环合得1。该路线的弊端在于:(1) 将氨基用光气或双光气异氰酸化得到异氰酸酯,其高度不饱和的基团对水分十分敏感,实验需严格控水,条件较苛刻;(2) 先接上侧链之后,以分子结构较大的中间体去成环,会使得反应副产物增多,影响总收率;(3)3-甲氨基-4,4,4-三氟巴豆酸乙酯的氮上甲基存在位阻效应,对成环有不利影响。
刘长令等报道了另一条合成路线:先用甲醇保护羧基,然后以较小的分子结构去合成脲嘧啶环中间体,最后连接侧链得1。但是该路线到目前为止没有实验报道。本文参考文献的思路,但改用氯甲酸乙酯和2-氯-5-氨基苯甲酸甲酯反应得2-氯-5-(乙氧羰基) 氨基苯甲酸甲酯(3);3与3-氨基-4,4,4-三氟巴豆酸乙酯环合得脲嘧啶环中间体(4); 4再经甲基化、水解、酰化和缩合反应合成1( Scheme 1),总收率55.2%,纯度99.1%。中间体和产物结构经1H NMR和EI-MS确证。
本文采用方法对4 的合成效果较好,既避免了异氰酸酯对水的敏感性,也消除了甲氨基的位阻效应,反应操作简便,收率较高。Scheme 1 合成路线具有操作方便、安全性好、收率较高等特点,适合工业化生产。
1 实验部分
1.1 仪器与试剂
ZF-7 型三用紫外分析仪; Brukor AVANCE400 MHz 型核磁共振仪(CDCl3为溶剂,TMS 为内标);HP 5989A 型质谱仪。
2-氯-5-硝基苯甲酸甲酯,2-羟基-2-甲基丙酸丙烯酯和3-氨基-4,4,4-三氟巴豆酸乙酯按文献方法合成;氢化钠( 含量60%),上海天莲化工有限公司;硫酸二甲酯,上海凌峰化学试剂有限公司;其余所用试剂均为分析纯。
1.2 合成
(1) 2-氯-5-氨基苯甲酸甲酯(2)的合成
在压力反应釜中加入2-氯-5-硝基苯甲酸甲酯20g(93mmol),1%Pt/C催化剂0.2g,三乙胺0.2g和甲醇40mL,搅拌下用氮气置换,于3.0MPa 氢压,85℃~90℃反应2h。抽滤,滤饼(催化剂)可重复使用。滤液加水50mL,用二氯甲烷(3×70mL) 萃取,合并萃取液,用饱和食盐水(3×50mL)洗涤,无水硫酸钠干燥,蒸除溶剂得淡黄色液体16.2g化合物2,收率93.9%。
(2) 3的合成
在四口烧瓶中依次加入9.3g(50mmol)化合物2,吡啶6.3g(80mmol)和二氯甲烷100mL,搅拌下于0℃~5℃滴加氯甲酸乙酯6.5g(60mmol),滴毕,于0℃~5℃反应30min;于室温反应3h(TLC跟踪)。加入二氯甲烷20mL,依次用1mol·L -1盐酸(3×30mL),饱和碳酸氢钠溶液(3×30mL)和水(3×30mL)洗涤,无水硫酸钠干燥,蒸除溶剂得黄色固体12.5g化合物3,收率97.0%。
(3) 2-氯-5-(1,2,3,6-四氢-3-甲基-2,6-二氧-4-三氟甲基嘧啶-1-基)苯甲酸甲酯(5)的合成
氮气保护,在四口烧瓶中加入氢化钠1.4g(35mmol)和无水DMF20mL,搅拌使其成悬浮液;加入N-甲基吡咯烷酮(NMP)3.0g(30mmol),于0℃~5℃缓慢滴加3-氨基-4,4,4-三氟巴豆酸乙酯5.5g(30mmol),滴毕,于40℃反应30min。于30 min内滴加37.7g(30mmol)的DMF(15mL) 溶液; 于室温反应30min; 于135℃~140℃反应5h。加入碳酸钾5.0g和硫酸二甲酯3.8g(30mmol),于室温反应3h。倒入30mL 氨水中,搅拌下加水50 mL,用乙酸乙酯(3×50mL) 萃取,合并萃取液,用饱和氯化钠溶液(3×50mL) 洗涤,无水硫酸钠干燥。抽滤,滤液脱溶后用异丙醇重结晶得白色固体,于60℃真空干燥12h得白色固体10.8g化合物5,m.p.167℃~168℃(168℃~169℃),收率80.9%(3→5)。
(4) 2-氯-5-(1,2,3,6-四氢-3-甲基-2,6-二氧-4-三氟甲基嘧啶-1-基)苯甲酸(6)的合成
在四口烧瓶中加入4.7g(13mmol)化合物5,冰醋酸30mL 和盐酸30mL,搅拌使其溶解;于70℃反应8h。减压蒸除冰醋酸,剩余物倒入100mL水中,有固体析出。过滤,滤饼用水洗至中性,于60℃ 真空干燥12h 得白色固体4.1g化合物6,收率91.2%。
(5) 1 的合成
在四口烧瓶中加入3.4g(10mmol)化合物6和氯仿30mL,搅拌使其溶解; 加入草酰氯1.8g(14mmol)和几滴DMF,于室温反应3h。过滤,滤液脱溶得棕色固体3.4g。加入氯仿20mL,搅拌使其溶解;加入吡啶1.5g 和少量碘化钠,加入2-羟基-2-甲基丙酸烯丙酯1. 6g(11mmol),于50℃~55℃反应5h(TLC跟踪)。倒入30mL水中,用二氯甲烷(3×20mL)萃取,合并萃取液,用1mol·L -1盐酸洗涤至pH5;用饱和碳酸氢钠溶液(3×20mL)洗涤,用水(3×20mL) 洗至中性,无水硫酸钠干燥。蒸除溶剂得油状物,用异丙醇重结晶得白色固体3.9g目的化合物1,m.p.110℃~112℃(113℃),收率82.2%。
2 结果与讨论
(1) 4的合成
合成脲嘧啶环结构是合成1 的关键反应。实验中,我们曾先在苯环上接上侧链基团制备异氰酸酯,再与3-甲氨基-4,4,4-三氟巴豆酸乙酯缩合成环。但TLC 跟踪发现,副产物较多,收率较低,而且带有侧链基团的取代苯胺与氯甲酸乙酯反应制备2-氯-5-(乙氧羰基氨基) 苯甲酸-1-烯丙氧羰基-1-甲基-乙基酯,将其与3-甲氨基-4,4,4-三氟巴豆酸乙酯缩合成环,TLC 跟踪显示,副产物也较多,收率较低。由此可见,侧链基团对于脲嘧啶环的形成是有影响的,并且甲氨基的位阻效应也增加了环合的困难。
在以上实验结果的基础上,我们采用2 与氯甲酸乙酯反应得3; 3 再与3-氨基-4,4,4-三氟巴豆酸乙酯缩合成环,并加入NMP 催化反应,环合后再用硫酸二甲酯甲基化,两步反应总收率较高(80.9%)。
(2) 2 的合成
文献方法采用铁粉还原硝基,虽然铁粉还原成本低,但后处理复杂,也严重污染环境。本文采用铂碳催化加氢的方式,收率较高(93.9%),纯度也较高(94.2%),后处理简单,且铂碳可以多次重复使用。
(3) 6 的合成
文献方法采用三溴化硼脱甲基制备6。由于三溴化硼具有较强的毒性和腐蚀性,遇水极易分解,在工业生产中存在诸多的安全问题。本文采用冰乙酸作溶剂,浓盐酸催化水解的方式,操作简便,安全性好,收率较高。
(4) 1 的合成
在1 的合成中,加入少量碘化钠催化反应。碘负离子具有较强的亲核性,同时又是很好的离去基团,缩短了反应时间,该反应与酰化反应的总收率为82.2%。同时,通过筛选溶剂,发现CHCl3作溶剂时反应的效果最佳,HPLC 跟踪发现纯度较高(99.1%),收率也较高(82.2%)。