非催化条件下异丙苯液相过氧化反应动力学
2015-08-21刘明鑫许志美孙伟振张明华赵玲
刘明鑫,许志美,孙伟振,张明华,赵玲
(华东理工大学化学工程联合国家重点实验室,上海 200237)
引 言
苯酚是重要化工原料及医学用品,主要用于生产酚醛树脂、己内酰胺、卤代酚、双酚A、烷基酚、水杨酸等[1-2]。丙酮也是重要的化工原料之一,可用于合成甲基丙烯酸甲酯、丙酮氰醇、环氧树脂、聚碳酸酯等化学品,也可用作溶剂、稀释剂和萃取 剂[3]。异丙苯法是目前联产苯酚与丙酮的主要方法[4],包括苯烷基化、异丙苯液相过氧化、过氧化氢异丙苯催化分解等过程,其中异丙苯过氧化是关键反应步骤[5]。为了提高反应速率,碱金属、过渡金属配合物、卟啉类化合物等被用来催化异丙苯过氧化[6-9]。然而,非催化条件下的异丙苯液相过氧化过程在未来相当长时间内仍将是工业异丙苯液相氧化的主导工艺。异丙苯液相过氧化属于链式自由基反应,反应过程中存在着大量自由基和中间产物,反应机理较为复杂,给动力学研究带来一定困难。
准确的反应动力学模型是进行化工过程优化、设计以及放大的基础[10-14]。Hattori 等[15]考虑氧气分压的影响,最早建立了异丙苯氧化动力学模型。于学敏等[16]认为氧气分压和空塔气速超过一定值时可以忽略气液两相中氧气的传质阻力。但是,上述研究将异丙苯过氧化的主要副产物α-甲基苄醇(MBA)和苯乙酮(ACP)合并,这样的处理方式使动力学模型过于简化而不能区分副产物。并且早期的异丙苯过氧化研究由于分析条件所限并未考虑链终止产物ROOR(DCP)的分解。然而,异丙苯过氧化中的DCP 在产率上与ACP 相当,也是一种重要的化工产品[17]。Andrigo 等[18]对异丙苯氧化动力学和反应机理进行过详细研究,但并没有给出动力学模型参数。Bhattacharya[19]集成前人研究成果,进一步总结了异丙苯氧化反应机理并提出了改进的动力学模型,但是其给出的动力学参数与其他文献存在较大差异并缺少实验数据的验证。
本文研究异丙苯液相过氧化反应动力学,着重考察副产物α-甲基苄醇(MBA)、苯乙酮(ACP)以及链终止产物ROOR(DCP)的生成。基于链式自由基反应机理,建立包含反应物异丙苯(IPB),主产物过氧化氢异丙苯(IPBHP),副产物MBA、ACP 以及链终止产物DCP 的反应动力学模型。
1 实验部分
1.1 实验装置及分析方法
异丙苯液相过氧化反应动力学实验装置如图1所示。反应釜为钛材,容积为1 L,采用油浴夹套加热。釜内配有搅拌器、挡板等内附件;釜外配有冷凝回流系统以防止反应液随尾气被带出体系。实验过程中反应温度的波动控制在±0.5℃之内。实验为半连续操作,每次实验一次性投入500 ml 原料异丙苯、2.1%过氧化氢异丙苯和1%稳定剂Na2CO3。氮气保护下搅拌升温,待温度升至设定温度后迅速将氮气切换为空气并开始计时。实验过程中每隔0.5~1 h 取样1 次。
反应混合液中的IPB、ACP 和MBA 釆用气相色谱法分析,内标法定量,内标物为三甲苯(TMB),分析柱为毛细管柱。程序升温:50℃保持4 min,后以10℃·min-1速率升温至90℃,继续以5℃·min-1速率升至120℃,最后以10℃·min-1速率升至280℃。主产物IPBHP 采用电位滴定碘量法分析;链终止产物DCP 采用高效液相色谱分析,以甲醇与 水按90:10 混合作为流动相。每组实验数据均为二次平行实验的平均值。图2和图3分别为反应混合物气、液相色谱图。

图1 异丙苯液相氧化实验装置Fig.1 Experimental setup of liquid-phase oxidation of isopropyl benzene
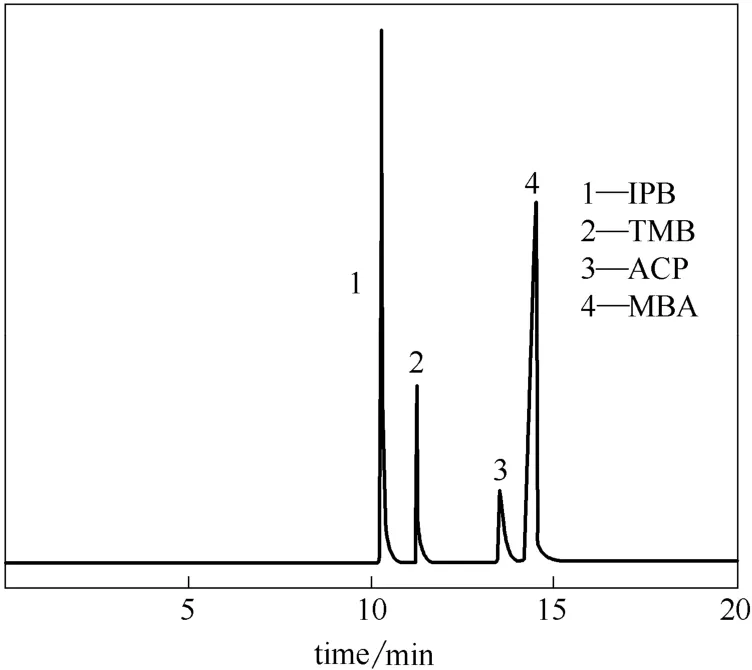
图2 异丙苯氧化产物气相色谱图Fig.2 Gas chromatogram of oxidation product of isopropyl benzene
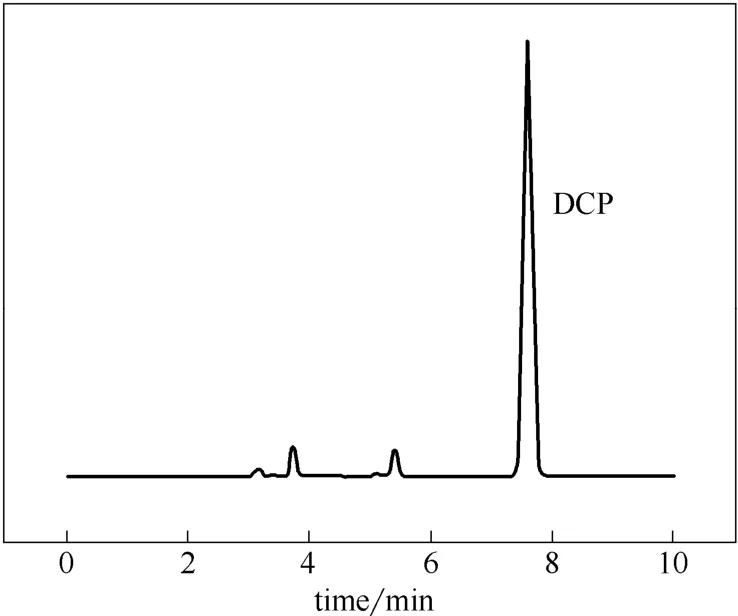
图3 DCP 的典型HPLC 谱图Fig.3 High performance liquid chromatogram (HPLC) of dicumyl peroxide (DCP)
1.2 预实验及实验条件
预实验结果表明,在通气量≥3 L·min-1,转速≥600 r·min-1时传质对反应的影响可以忽略。因此,后续实验均是在上述实验条件下进行以排除传质的影响。根据目前工业条件,选择373~404 K 4 个温度系列进行动力学实验,测得不同温度下液相各组分浓度随反应时间的变化。
2 反应机理及动力学模型建立
异丙苯液相过氧化属链式自由基反应,可分为3 个反应阶段,即链引发、链传递和链终止。Andrigo等[18],综合了Hattori 等[15]、Hendry[20]的研究结果得出了较为全面的反应机理[18,20]。Bhattacharya[19]认为Andrigo 等给出的某些基元反应步骤发生的概率很小,在保留链传递和交叉终止步骤的基础上,Bhattacharya 提出ROOH 分解产生的自由基是引发反应的主要因素,同时对 3CH ·、2RO ·、RO·与氧气结合过程提出了自己的看法。
链的引发方式有3 种,即引发剂引发、原料异丙苯引发、产物过氧化氢异丙苯引发
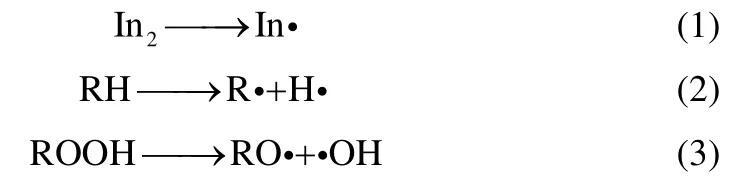
由于本实验是在氧气充足条件下进行的,所以在链终止阶段式(4)、式(5)两个基元反应不占主导地位,可忽略[17]

同时 CH3·会迅速与 O2反应生成,且在体系中RH 相对于其他组分大量存在,故有式(6)、式(7)两个反应发生
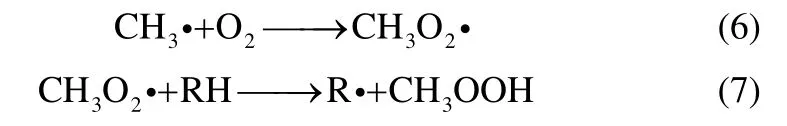
在反应过程中,除主产物外还存在其他中间产物。MBA 主要通过自由基RO·与RH 链传递反应生成,而ACP 则是通过RO·的β裂解生成[15,19],反应方程式为
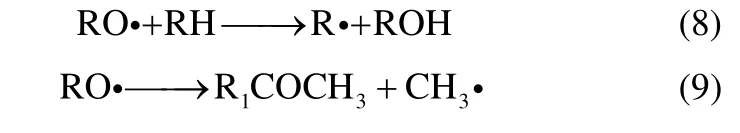
DCP 是反应过程中的链终止产物ROOR,通过式(10)生成
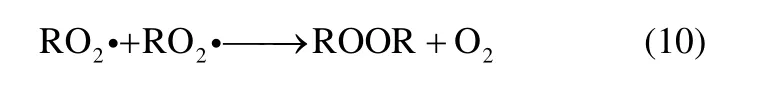
在对实验数据的分析中可以发现DCP 在反应开始持续一定时间后有下降的趋势,与IPBHP 的变化相似。说明当DCP 积累到一定浓度时分解成其他物质,然而大多数文献报道的反应机理中没有提及DCP 分解的基元反应。DCP 化学结构中含有不稳定的O—O 键和C—O 键,因此推测有式(11)、式(12)两种分解方式
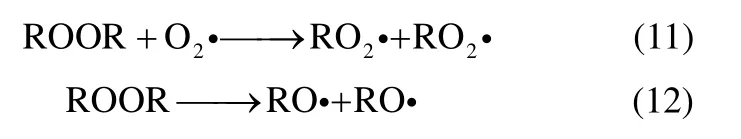
综上所述,氧气充足条件下异丙苯液相过氧化的关键基元反应步骤如下所示。
链引发
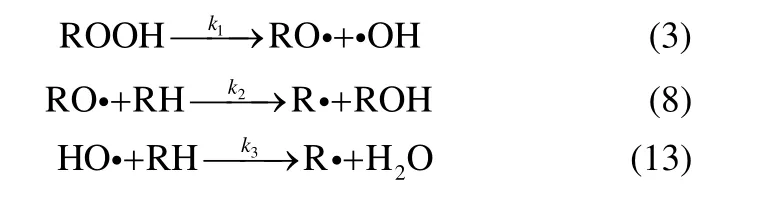
链传递
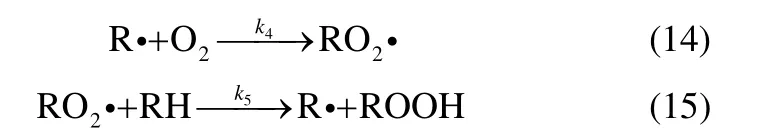
链终止

中间产物生成
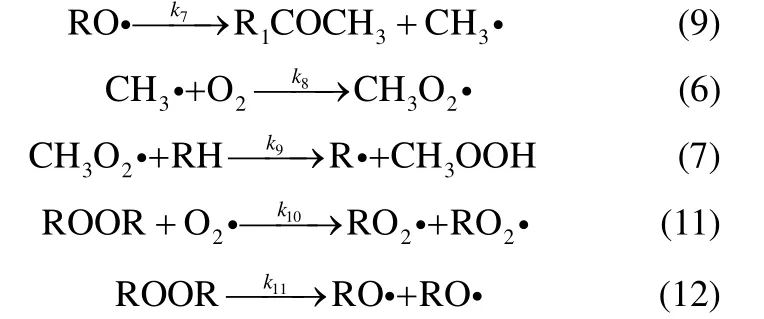
式中,R·、RH、ROOH、2RO ·、ROH、R1COCH3、ROOR 分别代表异丙苯基自由基、异丙苯、过氧化氢异丙苯、过氧化氢异丙苯基自由基、二甲基苄醇、苯乙酮、二甲基异丙苯。基于上述反应路径可以得出反应速率方程式(16)~式(25)
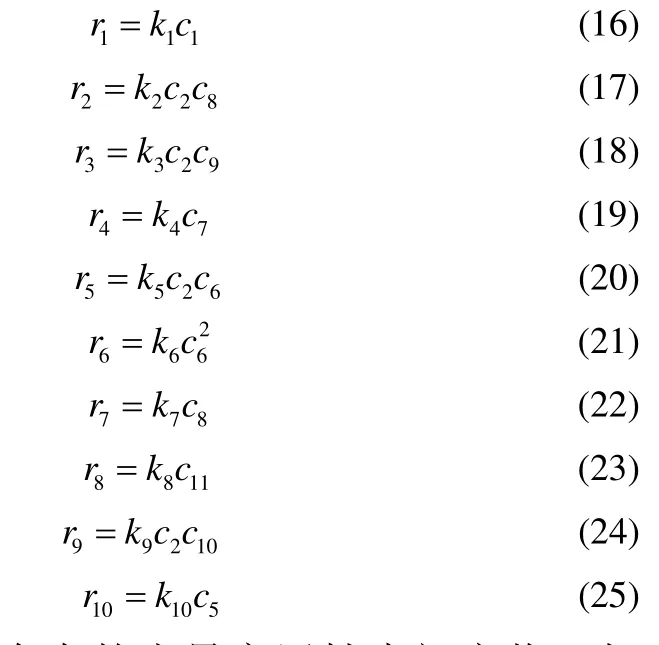
反应过程中存在的少量高活性中间产物,如·OH、CH3O2·、CH3·。初步的模拟计算发现这些自由基组分浓度在整个反应过程中基本为零;然而对于活性相对较低的自由基,如R·、2RO ·、RO·,模拟计算表明其累积速率不可忽视。因此,可将·OH、CH3O2·、C H3· 3 种自由基组分定义为高活性中间产物,对于这些高活性且浓度极小的中间产物,使用拟稳态假设使它们不在反应方程式中出现以达到模型简化的目的。类似的处理方法在文献中也可以看到[19]。
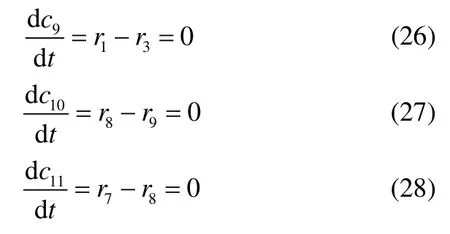
进一步,写出其他组分浓度随时间变化的常微分方程式(ODEs),并将式(26)~式(28)中的高活性组分浓度求解出并代入常微分方程组。整理后,可得出间歇条件下异丙苯液相过氧化动力学模型
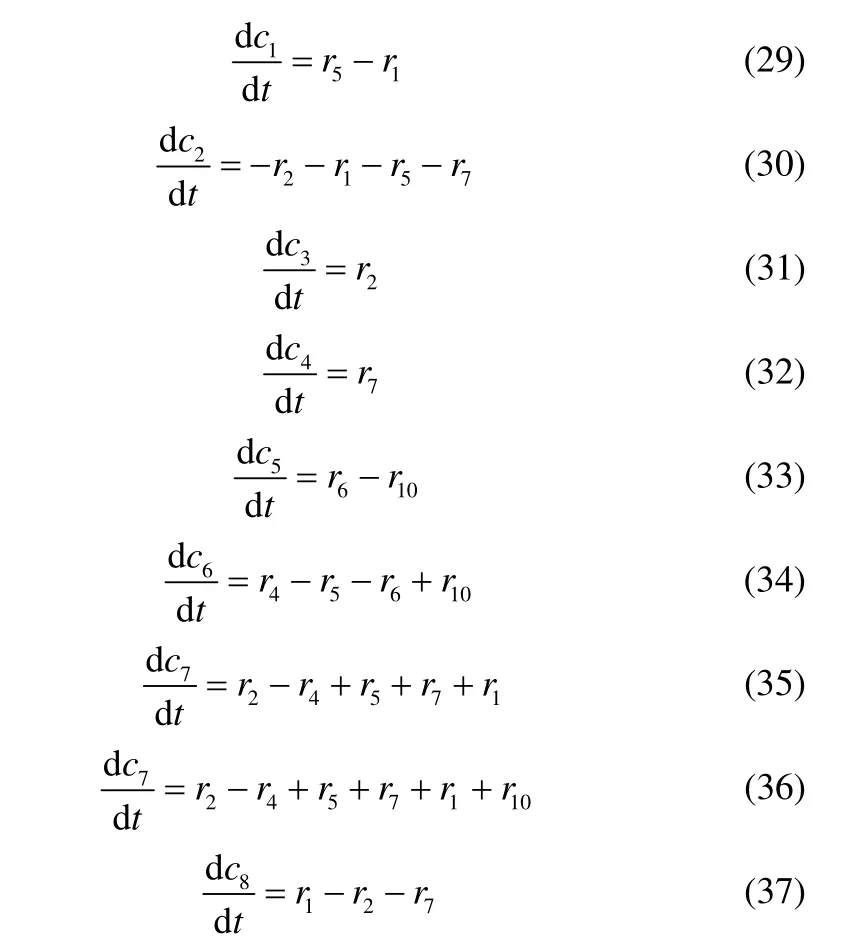
式中,下角标1 为ROOH;2 为RH;3 为ROH;4 为 R1C OCH3;5 为ROOR;6 为 RO2·;7 为R·;8 为RO·;9 为·O H;10 为 CH3O2·;11 为 CH3·;r1~r10为基元反应速率。上述常微分方程组的初始条件为,时间t=0时,c1=c10,c2=c20,c3=0,c4=0,c5=0,c6=0,c7=0,c8=0。
3 结果与讨论
已发表的异丙苯过氧化文献均未讨论氧化过程中链终止产物DCP 的分解。然而,对于大多数烃类氧化反应,链终止产物ROOR 在一定温度下易于发生进一步的分解[17]。本文假设存在以下3 种情况,即无ROOR 的分解反应发生(模型Ⅰ),ROOR 按照反应方程式(11)进行(模型Ⅱ);ROOR 按照反应方程式(12)发生(模型Ⅲ)。建立以上3 种情况下的动力学模型,并分别对404 K 下DCP 浓度随时间变化的实验数据进行拟合,拟合结果如图4所示。
由图4可以看出模型Ⅲ计算值远小于实验值。而模型Ⅰ即传统模型没有考虑DCP 的分解,导致虽前期较吻合,但当DCP 积累到一定浓度时没有表现出下降的趋势。模型Ⅱ的拟合值与实验值吻合较好,这表明在氧气充足条件下,溶解在芳烃环境中DCP的分解是按照式(11)进行的。
基于上述结论,利用4 个反应温度(373、383、393、404 K)下异丙苯过氧化过程中主副产物浓度随时间变化的实验数据,分别对异丙苯液相过氧化自由基模型中的参数进行拟合,拟合结果如图5所示。从图可以看出,低温条件下模型计算值与实验数据符合程度较好,而高温条件下拟合结果稍差,其原因可能是高温条件下中间反应较复杂,副产物较多,模型简化带来一些误差。
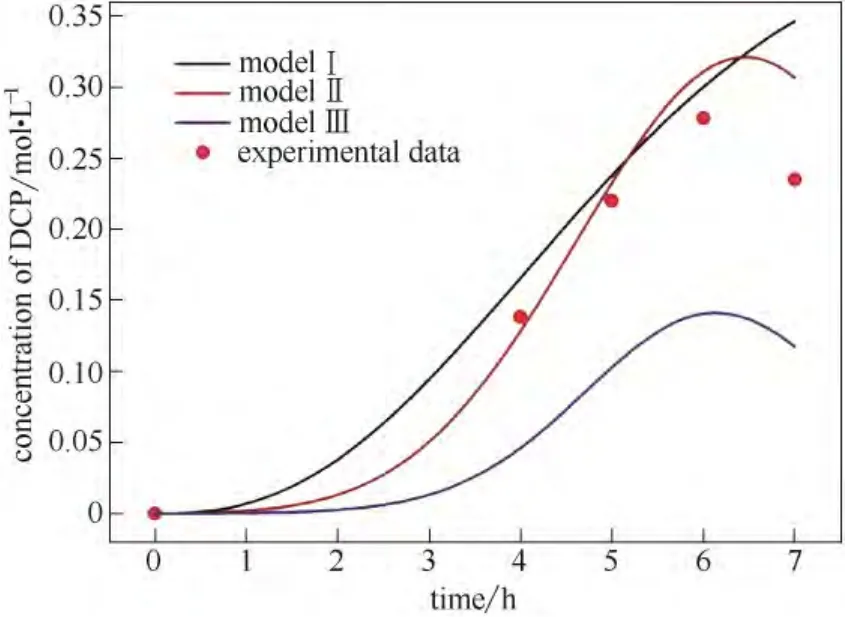
图4 404 K 下ROOR(DCP) 3 种分解途径模型拟合值 与实验值的比较Fig.4 Comparison between calculated and experimental data with different methods of DCP decomposition at 404 K
表1给出了拟合得到的各步反应动力学参数值,其中,k1~k10分别代表各步基元反应的速率常数,k4和k5分别代表链传递阶段的速率常数。同一温度下k4远大于k5,进一步证明了在氧气供给充足的条件下k5为链传递过程中的控制步骤。根据Arrhenius 关系式lnki与1/T的关系,计算得到各步反应的活化能依次为52.6(Ea,1)、189.6(Ea,2)、260.4(Ea,4)、49.0(Ea,5)、71.5(Ea,6)、171.5(Ea,7)、73.4 kJ·mol-1(Ea,10)。图6为各速率常数的Arrhenius关系。
图6表明lnki均随1/T的增大而线性递减,符合烃类氧化基元反应规律。Ea,2和Ea,7分别为副产物MBA 和ACP 的生成步骤所对应的活化能,Ea,5为主产物IPBHP 生成步骤对应的活化能。从数值上看,Ea,2和Ea,7分别为189.6 kJ·mol-1和171.5 kJ·mol-1,均大于Ea,5,说明同一条件下副产物的生成步骤对温度的敏感程度远大于主产物。这一结论与实验结果相符,也是工业上不盲目追求高温加快IPBHP 生产速率,而选择适当温度范围以提高选择性的主要原因。
4 结 论
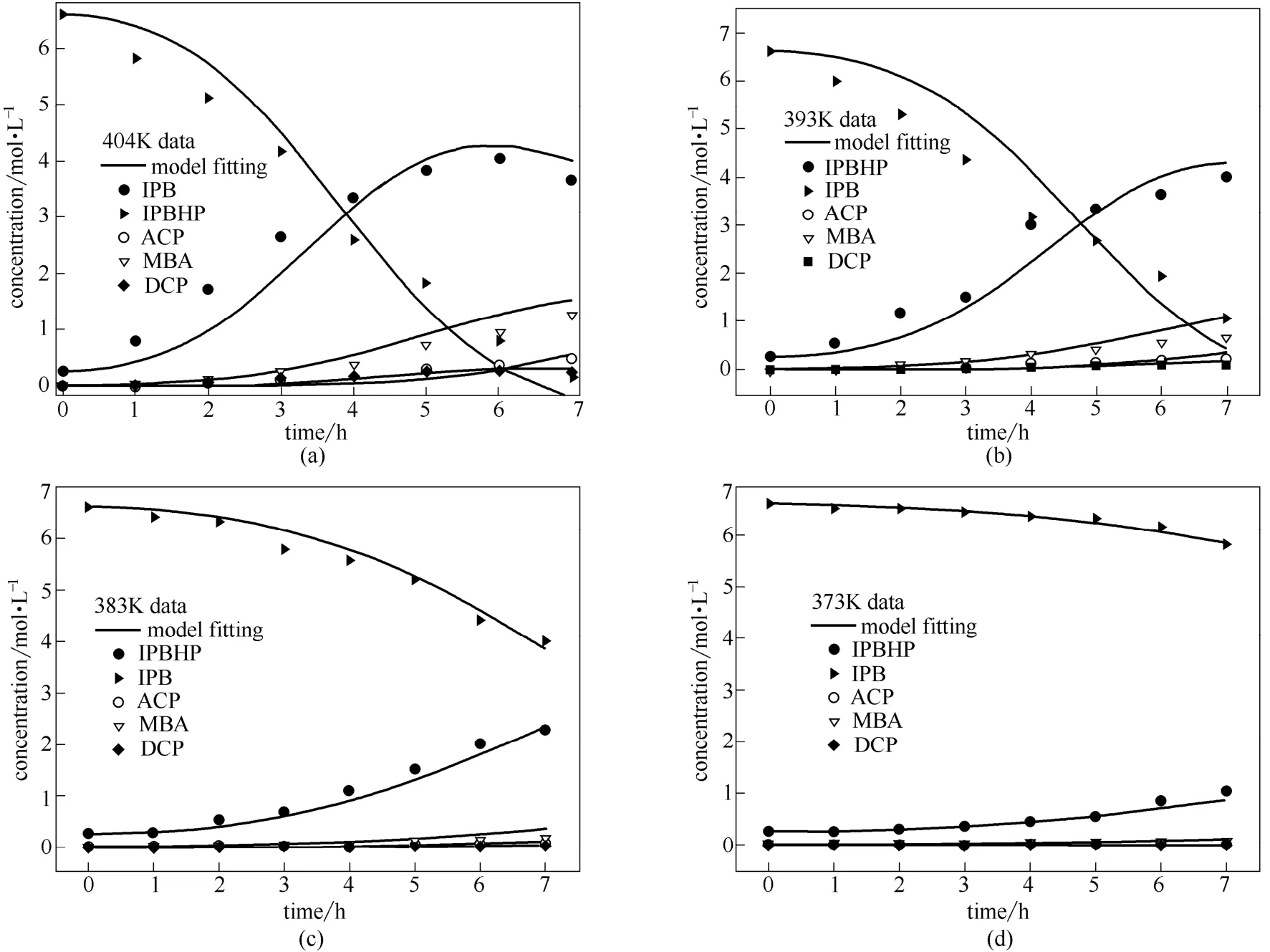
图5 不同温度条件下异丙苯液相氧化产物浓度分布Fig.5 Concentration of components in mixture at different temperatures
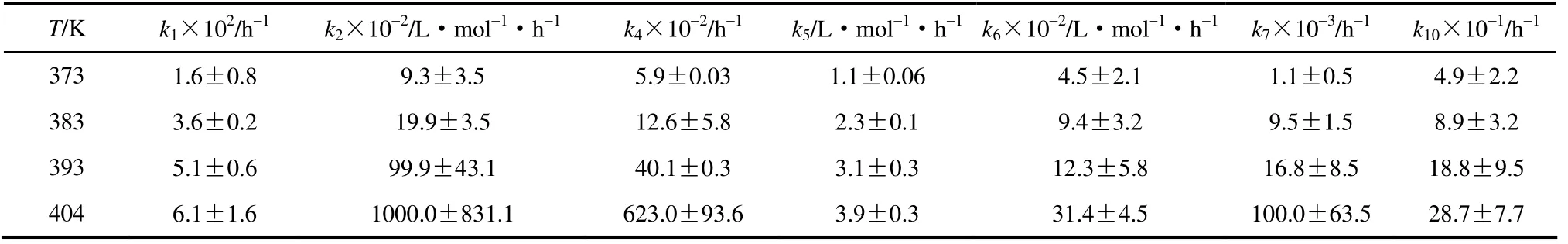
表1 不同温度下IPB 液相氧化速率常数估计及95%置信区间Table 1 Estimated rate constants with 95% confidence intervals
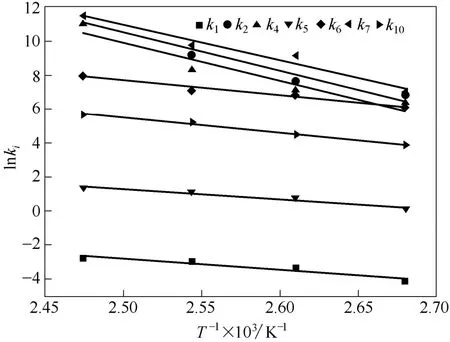
图6 Arrhenius 拟合结果Fig.6 T -1 and k calculated from Arrhenius fitting
实验在排除气液传质影响的条件下研究了非催化条件下异丙苯液相氧化的主副反应动力学。基于自由基链式反应机理建立了异丙苯氧化反应动力 学模型。本工作着重对链终止产物DCP 的生成及分解过程进行实验与模拟计算,证明了DCP 的分解是在氧气的参与下进行的。通过对373~404 K 下异丙苯氧化实验数据的拟合,得到了不同温度下各基元反应速率常数和活化能。
符 号 说 明
ki——基元反应速率常数,h-1或L·mol-1·h-1
ci——各物质浓度,mol·L-1
Ea——活化能,kJ·mol-1
[1]Schmidt R J.Industrial catalytic processes:phenol production [J].Applied Catalysis A:General,2005,280 (1):89-103
[2]Ren Yongli (任永利),Mi Zhentao (米振涛).Study on catalyst hydrogen peroxide oxidation of benzene into phenol [J].Chemical Industry and Engineering Progress(化工进展),2002,21 (11):827-830
[3]Han L,Wang Y,Zhang J,et al.Acidic montmorillonite/cordierite monolithic catalysts for cleavage of cumene hydroperoxide [J].Chinese Journal of Chemical Engineering,2014,22 (8):854-860
[4]Yadav G D,Asthana N S.Selective decomposition of cumene hydroperoxide into phenol and acetone by a novel cesium substituted heteropolyacid on clay [J].Applied Catalysis A:General,2003,244 (2):341-357
[5]He Yufeng,Wang Rongmin,Liu Yuyang,et al.Study on oxidation mechanism of cumene based on GC-MS analysis [J].Journal of Molecular Catalysis A:Chemical,2000,159 (1):109-113
[6]Crites C O L,Hallett-Tapley G L,Frenette M,et al.Insights into the mechanism of cumene peroxidation using supported gold and silver nanoparticles [J].ACS Catalysis,2013,3 (9):2062-2071
[7]Zhang Meiying,Wang Lefu,Ji Hongbing,et al.Cumene liquid oxidation to cumene hydroperoxide over CuO nanoparticle with molecular oxygen under mild condition [J].Journal of Natural Gas Chemistry,2007,16 (4):393-398
[8]Maksimov Y V,Suzdalev I P,Tsodikov M V,et al.Study of cumene oxidation over zirconia-,titania- and alumina-based complex oxides obtained by sol-gel methods:activity-structure relationships [J].Journal of Molecular Catalysis Chemical,1996,105 (3):167-173
[9]Wen Fei (文飞),Cheng Youwei (成有为),Guo Xia (郭霞),Wang Lijun (王丽军),Li Xi (李希).Liquid phase catalytic oxidation of cumene [J].Chemical Reaction Engineering and Technology(化学反应工程与工艺),2009,25 (4):376-379
[10]Sun Weizhen,Shi Yi,Chen Jie,et al.Alkylation kinetics of isobutane by C4olefins using sulfuric acid as catalyst [J].Industrial & Engineering Chemistry Research,2013,52 (44):15262-15269
[11]Sun Weizhen,Zhao Ling.Simulation of secondary oxidation ofp-xylene in liquid phase [J].Industrial & Engineering Chemistry Research,2010,50 (5):2548-2553
[12]Song J,Gao L,Lin J,et al.Kinetics and modeling of chemical leaching of sphalerite concentrate using ferric iron in a redox-controlled reactor [J].Chinese Journal of Chemical Engineering,2013,21 (8):933-936
[13]Sun Weizhen (孙伟振),Huang Huan (黄欢),Gu Xiaowu (顾晓吴),Zhao Ling (赵玲).Modeling of liquid phase oxidation kinetics of aromatic hydrocarbon based on free radical chain reaction mechanism [J].CIESC Journal(化工学报),2010,61 (7):2119-2123
[14]Xu Xiaotao (许晓涛),Zhang Weixing (张未星).Kinetics of 1,1,1,3,3-pentachloropropane synthesis reaction [J].CIESC Journal(化工学报),2014,65 (1):176-181
[15]Hattori K,Tanaka Y,Suzuki H,et al.Kinetics of liquid phase oxidation of cumene in bubble column [J].Journal of Chemical Engineering of Japan,1970,3 (1):72-78
[16]Yu Xuemin (于学敏),Li Ziming (李自明),Wang Jinquan (王金泉),et al.Study of cumene liquid-phase oxidation reaction kinetics [J].Journal of Chemical Industry and Engineering(China) (化工学报),1984,35 (2):130-138
[17]Di Somma I,Marotta R,Andreozzi R,et al.Dicumyl peroxide thermal decomposition in cumene:development of a kinetic model [J].Industrial & Engineering Chemistry Research,2011,51 (22):7493-7499
[18]Andrigo P,Caimi A,Cavalieri P,et al.Phenol-acetone process:cumene oxidation kinetics and industrial plant simulation [J].Chemical Engineering Science,1992,47 (9):2511-2516
[19]Bhattacharya A.Kinetic modeling of liquid phase autoxidation of cumene [J].Chemical Engineering Journal,2008,137 (2):308-319
[20]Hendry D G.Rate constants for oxidation of cumene [J].Journal of the American Chemical Society,1967,89 (21):5433-5438
