Visualizing the Ensemble Structures of Protein Complexes Using Chemical Cross-Linking Coupled with Mass Spectrometry
2015-08-08ZhouGongYueHeDingXuDongNaLiuErquanZhangMengQiuDongChunTangCASKeyLaboratoryofMagneticResonanceinBiologicalSystemsStateKeyLaboratoryofMagneticResonanceandAtomicMolecularPhysicsWuhanInstituteofPhysicsandMathematicsChi
Zhou Gong,Yue-He Ding,Xu Dong,Na Liu,E.Erquan Zhang,Meng-Qiu Dong✉,Chun Tang✉CAS Key Laboratory of Magnetic Resonance in Biological Systems,State Key Laboratory of Magnetic Resonance and Atomic Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences, Wuhan 43007,China
2National Institute of Biological Sciences,Beijing 102206,China
METHODS
Visualizing the Ensemble Structures of Protein Complexes Using Chemical Cross-Linking Coupled with Mass Spectrometry
Zhou Gong1,Yue-He Ding2,Xu Dong1,Na Liu2,E.Erquan Zhang2,Meng-Qiu Dong2✉,Chun Tang1✉1CAS Key Laboratory of Magnetic Resonance in Biological Systems,State Key Laboratory of Magnetic Resonance and Atomic Molecular Physics,Wuhan Institute of Physics and Mathematics,Chinese Academy of Sciences, Wuhan 430071,China
2National Institute of Biological Sciences,Beijing 102206,China
Received:23 October 2015/Accepted:11 November 2015/Published online:28 December 2015
Graphical Abstract

Chemical cross-linking coupled with mass spectrometry(CXMS)identifes protein residues that are close in space,and has been increasingly used for modeling the structures of protein complexes.Here we show that a single structure is usually suffcient to account for the intermolecular cross-links identified for a stablecomplex with sub-μmol/L binding affinity. In contrast, weshow that the distance between two cross-linked residuesin the different subunits of a transient or fleeting complexmay exceed the maximum length of the cross-linker used,and the cross-links cannot be fully accounted for with aunique complex structure. We further show that theseemingly incompatible cross-links identified with highconfidence arise from alternative modes of protein-proteininteractions. By converting the intermolecular crosslinksto ambiguous distance restraints, we established arigid-body simulated annealing refinement protocol toseek the minimum set of conformers collectively satisfyingthe CXMS data. Hence we demonstrate that CXMSallows the depiction of the ensemble structures of proteincomplexes and elucidates the interaction dynamics fortransient and fleeting complexes.
Protein-protein interaction,Encounter complex,Fleeting complex,Ensemble refnement, Ambiguous distance restraint
INTRODUCTION
A protein interacts with other proteins to perform its function.The binding affnity or KDvalue between two proteins ranges over ten orders of magnitude,and the resulting complex can be stable,transient or feeting (Jones and Thornton 1996;Nooren and Thornton 2003).Examples of stable complexes include enzyme/ enzyme inhibitor and antigen/antibody(Kastritis et al. 2011),while transient and feeting complexes are often involved in cell signaling.Transient complexes are those with KDvalues greater than 1 μmol/L, whereas feeting complexes are three-four orders of magnitude weaker with KDvalues in mmol/L(Vinogradova and Qin 2012;Xing et al.2014;Liu et al. 2016).
Two transiently interacting proteins not only form a stereospecifc complex,they can also form a series of nonspecifc encounter complexes(Tang et al.2006; Fawzi et al.2010;Schilder and Ubbink 2013).Encounter complexes are important structural intermediates,and facilitate the formation of the stereospecifc complex. Yet,encounter complexes constitute only a minor population of the total complex,and are diffcult to study (Berg et al.1981;Schreiber and Fersht 1996;Gabdoulline and Wade 2002).With the KDvalue in mmol/L, the distinction between specifc and non-specifc complexes starts to blur,and the subunits in a feeting complex often adopt a variety of conformations(Tang et al.2008;Liu et al.2012).As such,to characterize the structure of a protein complex,especially a transient or feeting complex,itoften requires an ensemble description to recapitulate the different conformational states.
Chemical cross-linking of proteins coupled with mass spectrometry analysis(CXMS)is an emerging technique to investigate protein-protein interactions (Rappsilber 2011;Herzog et al.2012;Kalisman et al. 2012;Lasker et al.2012;Walzthoeni et al.2013; Politis et al.2014).Amine-specific homo-bifunctionalcross-linkers, including bis-sulfosuccinimidyl suberate (BS3)and bis-sulfosuccinimidyl glutarate(BS2G),are commonly used.Recently,carboxylate-specifc crosslinkers reactive towards glutamate or aspartate residues,such as pimelic acid dihydrazide(PDH;Leitner et al.2014),were added to the CXMS toolbox.In theory,two primary amine groups(either lysine side chain orprotein N-terminus)ortwo carboxylate groups(either glutamate or aspartate side chains)that are close in space can be covalently linked.The crosslinked residues can be identifed with the use of a database search engine(Rinner et al.2008;Yang et al. 2012),and each intermolecular cross-link can be converted to a distance restraint for modeling the complex structure(Rappsilber 2011;Kalisman et al. 2012;Walzthoeni et al.2013;Schmidt and Robinson 2014).
As CXMS has been increasingly used for the structural characterization of protein complexes,two technical issues have become apparent(Rappsilber 2011; Merkley et al.2014).First,only a fraction of the crosslinks expected from the known structure of a protein complex are experimentally observed.This can be due to low accessibility and reactivity of the involved residues(Leitner et al.2014).Second and more intriguingly,for a subset of cross-links,the theoretical distance between two cross-linked residues,as calculated from the specifc complex structure,sometimes exceedsthe maximum length ofthe cross-linker (Kahraman et al.2013).Incorrect identifcation of cross-linked peptides has been blamed for such discrepancies(Zheng et al.2011;Kalisman et al.2012). Yet,with the most stringent criteria that essentially eliminate false identifcations,sometimes there remain cross-links violating the distance limits(Lossl et al. 2014).So what are the origins of these‘‘incompatible’’cross-links?
CXMS data have been recently implemented in ROSETTA software package for modeling protein complex structures(Kahraman et al.2013;Lossl et al. 2014).The approach aims to obtain a single structure that satisfes CXMS restraints and has the lowest ROSETTA energy score,and is suited for characterizing stable complex structures.Nevertheless,as transient and feeting complexes can adopt a multitude of conformational states,a single-conformation representation may not suffce.Here we show that the highly reliable but seemingly incompatible cross-links arise from alternative modes of protein-protein interactions.We present a rigid-body refnement protocol against all the experimental cross-links,and show that an ensemble representation comprising multiple conformers of the complex is often required when characterizing transient and feeting complexes.
RESULTS
Refnement of the stable complex structure
To refne against intermolecular CXMS restraints,we treated each subunit as a rigid body.Any two crosslinked lysine residues were restrained to have their Cα-Cαdistance to be less than the maximum length of the corresponding cross-linker using a square-well pseudo-energy potential.BS3and BS2G covalently link lysine residues<24 Å and<20 Å apart,respectively,as measured from Cαto Cαatoms(Lee 2009;Kahraman et al.2011).Cross-links may also involve protein N-terminus;when fully extended,the maximum Cα-Cαdistance between an N-terminal residue and a lysine is 15 Å for BS2G and 19 Å for BS3.
We then assessed the refnement protocol on the complex between trypsin and bovine pancreatic trypsin inhibitor(BPTI),a stable complex with a KDvalue of~60 fmol/L(Marquart et al.1983;Kastritis et al. 2011).Based on the known structure of the complex (PDB code 2PTC),there can be a maximum of 17 theoretical inter-subunit lysine-lysine cross-links with BS3cross-linking reagent(Table S1).Starting from the structures for the free proteins(PDB codes 4GUX and 1JV8,for trypsin and BPTI,respectively),we fxed the coordinates of trypsin and allowed BPTI to freely rotate and translate as a rigid body.With simulated annealing,we refned the complex structure against the CXMS restraints,with additional van der Waals repulsive term employed.Calculating one structure takes less than 2 min on a single core of Intel Xenon 5620 CPU.Repeating the calculation from different starting positions for the two subunits afforded a set of highly converged structures with overall root-mean-square deviation(RMSD)for backbone heavy atoms almost 0 Å.Importantly,the RMS difference between the CXMS model and the crystal structure was only 0.54 Å (Fig.1).
Further assessment of the rigid-body refnement protocol
In practice,however,it is rare to have as many as 17 intermolecular cross-links for a complex with the size of trypsin/BPTI(281 residues total and 18 lysine residues).Often,only a few cross-links can be experientially identifed.To assess how robust the refnement protocol is with fewer CXMS restraints,we obtained CXMS data from the published studies(Herzog et al.2012;Kahraman et al.2013)for the complex between protein phosphatase 2A catalytic subunit (PP2Ac) and immunoglobulin binding protein 1(IGBP1).PP2Ac and IGBP1 interact with each other with a KDvalue of~300 nmol/L(Jiang et al.2013),and six intermolecular cross-links were identifed between Lys28-Lys158, Lys33-Lys166,Lys35-Lys163,Lys40-Lys158,Lys40-Lys163, and Lys40-Lys166(from PP2Ac to IGBP1)(Herzog et al. 2012).Starting from the structures for free PP2Ac(PDB code 2NYL)and IGBP1(PDB code 3QC1)proteins,we obtained their complex structures by refning against the CXMS distance restraints.The probabilistic distribution was computed for PP2Ac with respect to IGBP1 in all the structural models and was shown as atomic probability map(Schwieters and Clore 2002),which encompassed the known complex structure(Fig.2A). Importantly,the overall backbone RMS difference between the CXMS models and the crystal structure for PP2Ac/IGBP1 complex was as small as 2.8 Å(Fig.2B) (Jiang et al.2013).
Then what is the minimum number of intermolecular cross-links needed to model the complex structure? With the use of three experimental cross-links involving PP2Ac Lys40(Lys40-Lys158,Lys40-Lys163,and Lys40-Lys166),the resulting structures took up similar positions(Fig.S1A)as the structures calculated using the full set of CXMS restraints,though a bit more scattered. With only one CXMS restraint,for example from PP2Ac Lys35to IGBP1 Lys163,the modeling still afforded a set of CXMS models that are similar to those calculated with the full set of experimental CXMS restraints(Fig.S1B). Thus,the more CXMS restraints were incorporated,the more converged the resulting models were.We also performed the structural refnement using fve out of the six cross-links,and then back-calculated the Cα-Cαdistance for the unused cross-link.Except for the crosslink between PP2Ac Lys28and IGBP1 Lys158,the calculated distances are mostly within the maximum length stipulated by the corresponding cross-linker(Table S2). Thus,the cross-link between PP2Ac Lys28and IGBP1 Lys158afforded a key restraint about the complex structure,and owing to the sparsity of the intermolecular cross-links,this cross-link is not redundantly provided by other cross-links.
Using CXMS,we characterized the complex between CDK9 and Cyclin-T1.This complex is responsible for transcription elongation,and its two subunits interact with each other at a KDvalue of~300 nmol/L (Baumli et al.2008).We focused our attention on the intermolecular cross-links that were identifed twice or more,for which the probability of being observed by random chance was below 10-8for at least one instance and below 10-3for additional instances(a false discovery rate cutoff of 0.05,an E-value cutoffrate of 10-3,spectral count≥2,and the best E-value cutoff of 10-8).With these stringent criteria,it would be unlikely that the cross-links were identifed by random chance,and the remaining cross-links should be correctly assigned.Three intermolecular cross-links were identifed for CDK9/Cyclin-T1(Table 1)and the corresponding MS2 spectra are shown in Fig.S2.For each,the two linked lysine residues were found within the maximum length of the cross-linker,as calculated from the known structure of the complex(Baumli et al.2008).

Fig.1 Comparison between the CXMS model and the X-ray structure for the complex between trypsin and BPTI.The two structures are superimposed by trypsin(orange cartoon),and BPTI in the CXMS model and in the crystal structure(PDB code 2PTC)are colored gray and blue,respectively.The CXMS model was obtained by refning against 17 theoretical inter-molecular cross-links.The RMS difference of backbone heavy atoms between the two complex structures is 0.54 Å.Lysine residues involved are labeled

Fig.2 CXMS model obtained for the complex between PP2Ac and IGBP1.A The distribution of PP2Ac with respect to IGBP1(orange cartoon)is shown as atomic probability map,plotted at 30%threshold and shown as gray meshes.B The RMS difference between the CXMS model(gray cartoon for PP2Ac)and the crystal structure of the complex(PDB code 4IYP)can be as small as 2.8 Å.With PP2Ac superimposed,IGBP1 in the crystal structure is shown as blue cartoon.Cross-linked lysine residues are labeled and the intermolecular cross-links are shown as red lines
We treated each subunit in CDK9/Cyclin-T1 as a rigid body,and refned against the intermolecular CXMS distance restraints:two cross-linked lysine residues were restrained to have their Cα-Cαdistance to be less than the maximum length of the corresponding cross-linkerusing a square-wellenergy potential.Since each intermolecular cross-link was observed with both BS2G and BS3cross-linkers (Table 1),we restrained the Cα-Cαdistance to be shorter than the length of BS2G(20 Å for lysine-lysine cross-links and 15 Å for lysine-protein N terminus cross-links).In the refnement,the coordinates for one subunit,CDK9,were fxed,while the other subunit, Cyclin-T1,was grouped as a rigid body,given full translational and rotational freedoms.A single intermolecular CXMS restraint was readily satisfed,but theresulting complex model was poorly converged,with Cyclin-T1 dangling along one side of CDK9(Fig.S3).As Lys74and Lys144are adjacent to each other in CDK9, cross-links of Cyclin-T1 Lys6to these two residues provided redundant information about the complex structure.Cyclin-T1 Lys100and CDK9 Lys56are located at the other side of the complex;as a result,the refnement against the corresponding cross-link restraint afforded a different but overlapping distribution of the complex.With all three restraints used,a narrower distribution was obtained(Fig.3A).Signifcantly,the structural models based on CXMS restraints encompassed the known crystal structure of CDK9/ Cyclin-T1,and the pairwise RMS difference between the CXMS model and the PDB structure was as small as 2.86 Å(Fig.3B).Thus,we show that the CDK9/Cyclin-T1 complex can be modeled as a single conformer, based on sparse CXMS distance restraints.
CXMS analyses of transient and feeting complexes
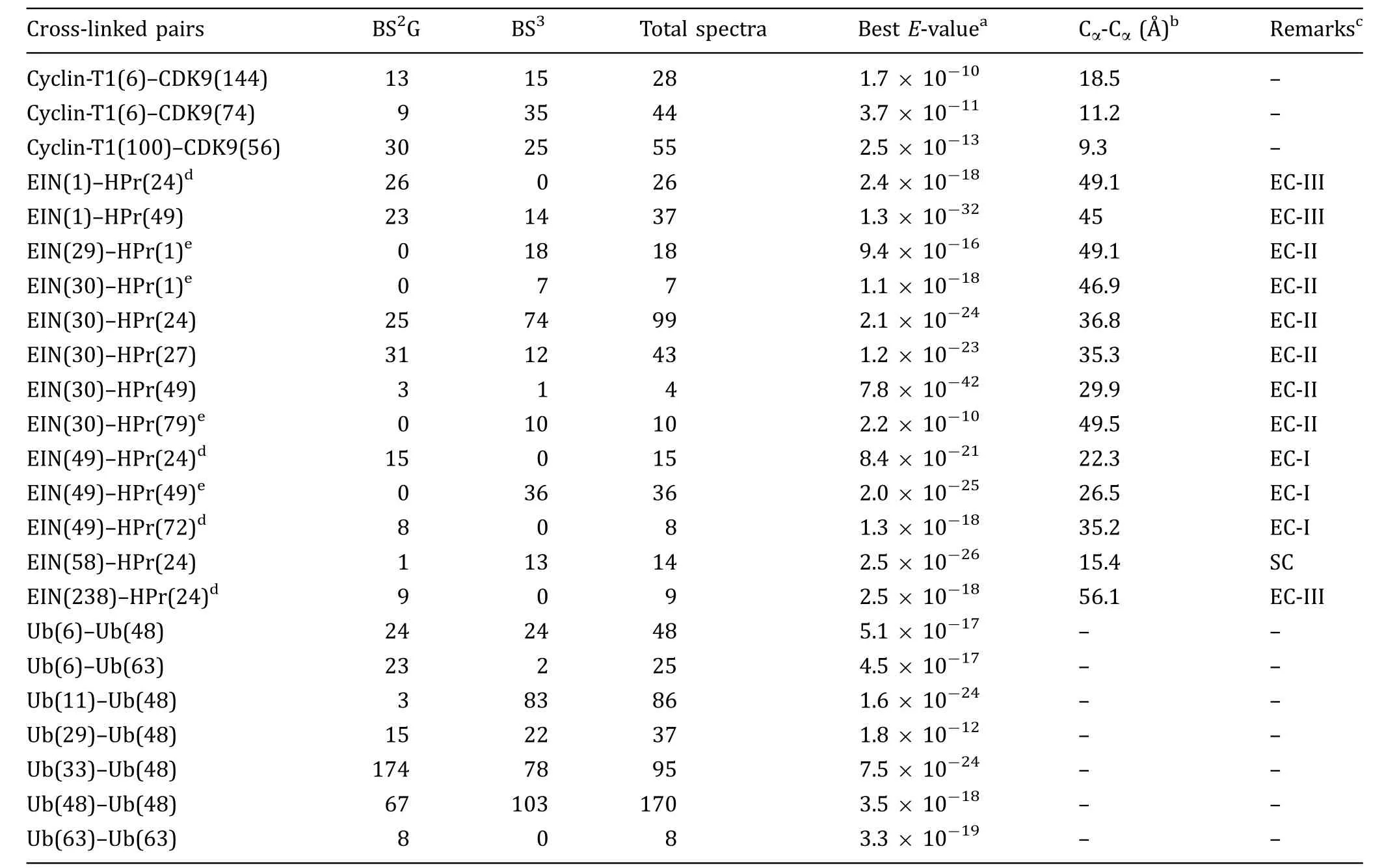
Table 1 Intermolecular cross-links observed for transient and feeting protein complexes
We then performed CXMS analysis for EIN/HPr and ubiquitin homodimeric complexes using BS2G and BS3. EIN and HPr are involved in signal transduction for bacterial sugar uptake and interact with each other with a KDvalue of~7 μmol/L(Suh et al.2007).Ubiquitin is an important signaling protein in cell and can noncovalently dimerize with a KDvalue of~5 mmol/L(Liu et al.2012).Using the same stringent criteria described above,intermolecular cross-links for the two complexes are also presented in Table 1,and the corresponding MS2 spectra are shown in Figs.S4 and S5.A total of 13 intermolecular cross-links were identifed for EIN/HPr, but only one of them(EIN Lys58to HPr Lys24)was found consistent with the stereospecifc complex structure (Garrett et al.1999).For validation,we also performedCXMS analysis for EIN/HPr using PDH(Leitner et al. 2014)as the cross-linking reagent.
In order to identify intermolecular cross-links between two ubiquitin subunits in a ubiquitin homodimer,we performed CXMS analysis on a mixture of14N-labeled(natural isotope abundance)and15N-labeled ubiquitin proteins(Liu et al.2012).The cross-links between14N-and15N-labeled peptides with characteristic MS1 spectra(Fig.S6)should only arise from intermolecular interactions(Taverner et al.2002).In this way,we identifed a total of seven intermolecular cross-links for the ubiquitin homodimer.
Ensemble structure refnement of protein encounter complexes
To account for the experimental cross-links and to model the structure of EIN/HPr complex,we fxed the position of EIN and treated HPr as a rigid body given rotational and translational freedoms.The intermolecular crosslinks could not be satisfed with a single-conformer representation of the complex,as the restraints were consistently violated with an average violation>8 Å (Fig.4A).This means that in addition to the stereospecifc complex,HPr sampled a multitude of conformations with respect to EIN,which were captured by cross-linking.Thus,weinvoked ensemble representation for the complex-with EIN fxed,HPr was represented as multiple conformers.We treated each intermolecular cross-link as an ambiguous restraint(Nilges 1995),and defned the CXMS energy averaged over all the conformers in the ensemble with a steep dependence on the Cα-Cαdistance.In this way,a CXMS restraint could be satisfed providing that it was accounted for by at least one conformer in the ensemble.The ensemble refnement showed that a minimum of four conformers was required to fully satisfy the intermolecular CXMS restraints with an average distance violation close to 0 Å (Fig.4A).Too large an ensemble size,however,would lead to over-ftting.When using fve conformers to represent the complex,HPr in the additional conformers were found scattering around,making no contribution to the CXMS energy(Fig.S7).
Using a spherical coordinate system,we projected the positions of HPr with respect to EIN in the CXMS models to lower dimensions.In the 2D plot,HPr was found in four distinct clusters(Fig.4B),thus explaining the requirement of four conformers in the ensemble.One cluster(SC)contained conformers overlapping with the known complex structure,and therefore accounted for the stereospecifc EIN/HPr interactions.HPr was positioned away from the specifc interface with EIN in the other three clusters(EC-I,EC-II and EC-III),which represented non-specifc interactions between EIN and HPr. Each cluster of conformers accounted for multiple intermolecular cross-links(Table 1).
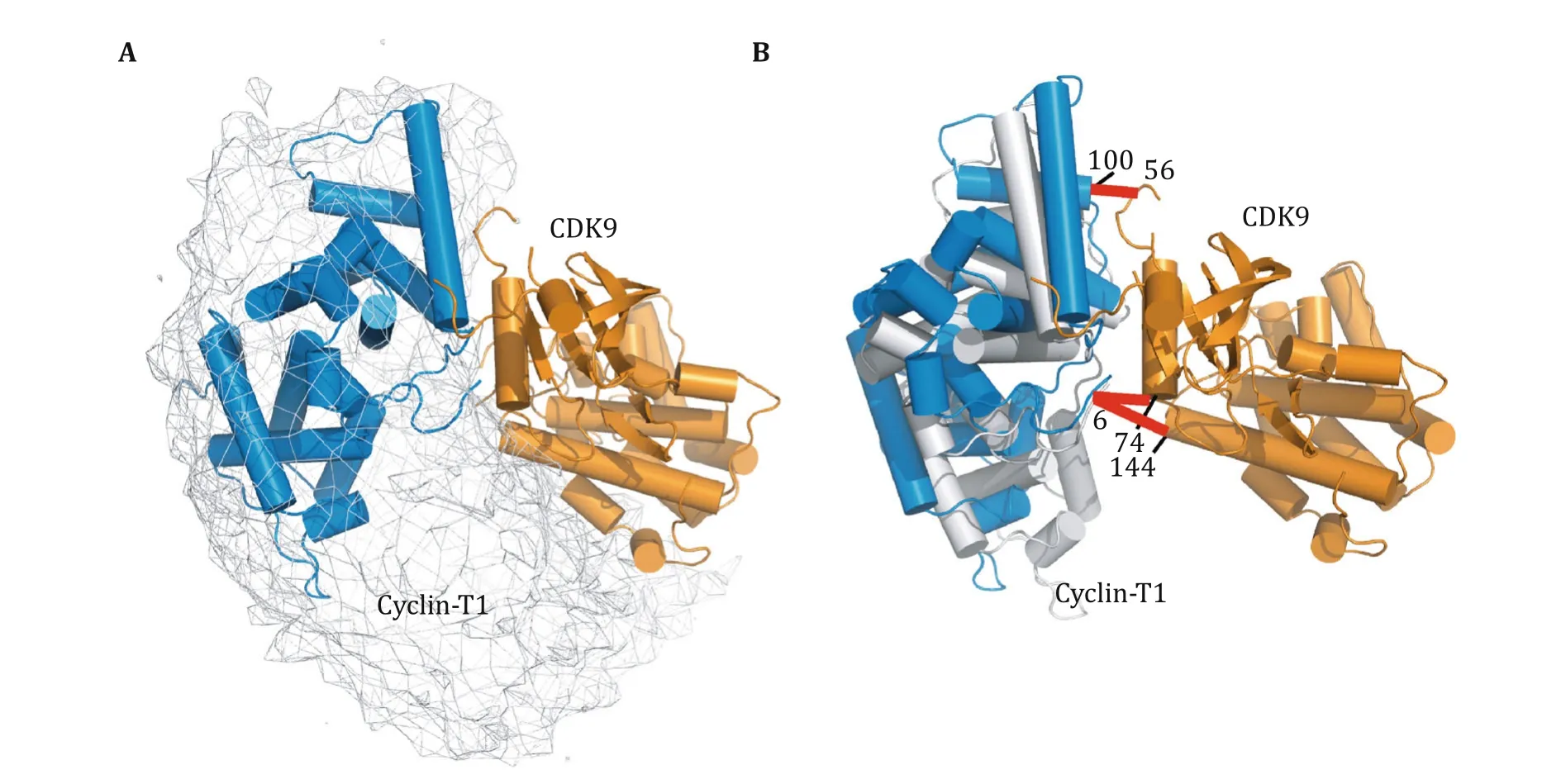
Fig.3 Structural model for the CDK9/Cyclin-T1 complex refned against intermolecular CXMS restraints.A The distribution of Cyclin-T1 with respect to CDK9(orange cartoon)is represented as an atomic probability map plotted at a 10%threshold(gray mesh).B A selected CXMS model,shown as orange and gray cartoon for CDK9 and Cyclin-T1,respectively.For comparison,the CDK9 of the crystal structure (PDB code 3BLH)is superimposed,and the Cyclin-T1 crystal structure is shown as a blue cartoon.The root-mean-square deviation between the two complex structures is 2.86 Å.Each set of two cross-linked residues is denoted with a red bar
We could cross-validate the ensemble structure modeled from lysine-lysine cross-links with the CXMS restraints from a different cross-linking reagent,PDH (Leitner et al.2014).For a pair of PDH cross-linked glutamate residues,the Cα-Cαdistance should be less than 22 Å.With high confdence,the PDH cross-links were identifed between EIN Glu41and HPr Glu85and between EIN Glu67and HPr Glu85(Fig.S8).Calculated from the stereospecifc complex structure(Garrett et al. 1999),the Cα-Cαdistances for these two pairs of residues were 41.2 and 12.9 Å,respectively.Clearly,the cross-link between EIN Glu41and HPr Glu85could not be accounted for with the stereospecifc complex structure alone.In the four-conformer ensemble structure modeled from BS2G/BS3CXMS data,however,the averaged Cα-Cαdistance between EIN Glu41and HPr Glu85was 23.1±4.9 Å.

Fig.4 Ensemble refnement for the complex structure between EIN and HPr.A Average violation of CXMS distance restraint(blue axis on the left)and the number of the satisfed restraints(orange axis on the right)versus the number of conformers representing the complex. With four or more conformers,all CXMS restraints can be satisfed.B Spherical coordinates for the four-conformer ensemble structures showing the distribution of HPr with respect to EIN.In each ensemble structure,the HPr is found in four clusters,namely EC-I,EC-II,ECIII,and SC.For comparison,the structure for EIN/HPr stereospecifc complex(PDB code 3EZA)is indicated as a cyan dot.C Atomic probability map of the distribution of HPr with respect to EIN in the ensemble structure refned against intermolecular CXMS restraints. The difference clusters of CXMS conformers are labeled.D Atomic probability map of the distribution of HPr with respect to EIN in the ensemble structure refned against intermolecular PRE data.The NMR ensemble was calculated based on the previously published data (Tang et al.2006).EIN is fxed and shown as orange cartoon,the distribution of HPr is shown as gray meshes and plotted at 20% threshold.For comparison,the stereospecifc complex structure is superimposed,with HPr shown as blue cartoon,and the four clusters are also marked
Previously,EIN/HPr complex has been characterized with paramagnetic nuclear magnetic resonance(NMR), and it was shown that EIN and HPr form a multitude of encounter complexes,which facilitate the formation of the stereospecifc complex(Tang et al.2006;Fawzi et al. 2010).Protein encounter complexes are of low occupancies and short lifetimes.Previous NMR studiesestimated that encounter complexes made up less than 10%of the total EIN/HPr complex,thus putting the apparent KDvalue for the encounter interactions>10 mmol/L(Fawzi et al.2010).Importantly,the distribution of HPr relative to EIN modeled on the basis of CXMS data(Fig.4C)resembles the EIN/HPr encounter complexes previously depicted using NMR spectroscopy (Fig.4D).
Ensemble structure refnement of a feeting complex
Performing CXMS experiments on an equimolar mixture of15N-and14N-labeled ubiquitin proteins,we identifed fve inter-molecular cross-links.We fxed the coordinates for one ubiquitin,and allowed the other one to move.A single conformation for the ubiquitin dimer failed to satisfy all the restraints,with average violations~2 Å.Hence we represented the ubiquitin dimer with two,three,and four conformers,with C2non-crystallographic symmetry enforced for each pair of ubiquitin dimer.The CXMS restraints could be satisfed with an N=2 ensemble.Increasing the size of the ensemble did not improve the agreement between experimental and calculated Cα-Cαdistances,and the additional conformers in the N=3 and 4 ensemble scattered around with respect to its dimer partner (Fig.S9).Thus,the N=2 ensemble was suffcient to describe the dynamic interactions between two ubiquitin proteins.
In the CXMS models,the two ubiquitins adopt a variety of orientations(Fig.5A),characteristic of feeting protein-protein interactions(Liu et al.2016).This also explains why Lys48in one ubiquitin was able to cross-link to fve different lysine residues,except for Lys27and Lys63,in the other ubiquitin.Importantly,the two subunits interacted at the β-sheet region in the CXMS models,and the distribution of the CXMS models was in good agreement with a previous NMR characterization of the ubiquitin homodimer(Fig.5B).
DISCUSSION
CXMS hasbeen increasingly used to characterize protein-protein interactions and to model protein complex structures(Walzthoeni et al.2013;Schmidt and Robinson 2014).However,when experimental cross-links cannot be accounted for with a unique structure,previous CXMS applications generally ignored‘‘incompatible’’ones or relaxed the Cα-Cαdistance restraints(Herzog et al.2012;Politis et al.2014).Here we show that CXMS is exquisitely sensitive to encounter and feeting protein-protein interactions that have apparent KDvalues in mmol/L,and those seemingly incompatible cross-links contain the information about the dynamics of protein-protein interactions.

Fig.5 Ensemble structure for the ubiquitin homodimer.With one ubiquitin subunit fxed(orange cartoon),the probabilistic distribution of the other ubiquitin subunit in the dimer is plotted at 20% threshold (gray meshes).The ensemble structures of ubiquitin homodimer were calculated by refning against A intermolecular CXMS restraints or B intermolecular NMR restraints
To account for the intermolecular cross-links identifed with high confdence,we established a rigid-body refnement protocol.The protocol enabled the depiction of the relative subunit distributions in a complex.We frst show that the refnement protocol can model the structures of stable complexes to high precision and accuracy.For transient and feeting ones,however,when a single conformation failed to satisfy all the intermolecular cross-links,we invoked ambiguous distance restraints,in which a distance restraint was accounted for by any one of the conformers in the ensemble (Fig.S10).Demonstrated with EIN/HPr and ubiquitin homodimeric complexes,we showed that the resulting structures satisfed the experimental intermolecular cross-links and recapitulated alternative modes of protein-protein interactions.Moreover,the lysine-and carboxylate-specifc cross-links for the EIN/HPr complex corroborate each other,which attests the power of CXMS in revealing the dynamics in protein interactions. Nevertheless,it should be noted that,though a qualitative validation of the ensemble structure can be readily performed,a complete cross-validation may not be feasible owing to the sparsity of the CXMS restraints.
Protein interaction dynamicshavebeen mostly characterized using NMR spectroscopy.Though NMR afforded more structural details than CXMS does,it only works for relatively small protein complexes and requires a large amount of isotopically labeled proteins. In contrast,CXMS is not limited by the size of the proteins,and can be performed on μg or ng of proteins of natural isotope abundance.CXMS is often used conjunction with other techniques like electron microscopy(EM;Rappsilber 2011;Thalassinos et al.2013).Nevertheless,the data from other technique are sometimes at odds with the CXMS data(Plaschka et al.2015).Since proteins dynamically interact with each other,we envision that the ensemble refnement protocol presented herein will allow the reconciliation of different types of data and enable the characterization of subunit rearrangement in these large complexes.The method described herein does not take into account the fexibility of each subunit.Yet we anticipate that CXMS would allow the visualization of the dynamics for each individual protein,providing that a large number of intra-molecular cross-links of high confidence areidentified using cross-linking reagents of differentlengths and chemical properties.
MATERIALS AND METHODS
Cross-linking reaction and analysis
CDK9,Cyclin-T1,EIN,HPr,and ubiquitin proteins were purifed as previously described(Garrett et al.1999; Baumli et al.2008;Liu et al.2012).To prepare15N-labeled protein,bacterial cells expressing ubiquitin were grown in M9 minimum medium with U-15NH4Cl as the sole nitrogen source.The two subunits in each complex were mixed at a 1:1 ratio-0.6 μmol/L for CDK9/Cyclin-T1,16 μmol/L for EIN/HPr and 70 μmol/L for the ubiquitin homodimer.Cross-linking reactions were performed at room temperature in 20 mmol/L HEPES buffer(pH 8.0,7.2 and 7.5 for CDK9/Cyclin-T1, EIN/HPr and ubiquitin, respectively) containing 150 mmol/L NaCl and 0.5 mmol/L BS3(Thermo Scientifc)or BS2G(Thermo Scientifc)for 1 h,and were quenched with 20 mmol/L NH4HCO3.Cross-linking reactions using PDH for EIN/HPr complex were performed at 37°C in 20 mmol/L HEPES buffer pH 7.2 containing 150 mmol/L NaCl and 11 mmol/L 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride for 1 h,and were quenched with 20 mmol/L NH4HCO3.The proteins were subsequently precipitated with ice-cold acetone,air dried,and resuspended in 8 mol/L urea,100 mmol/LTris pH 8.5.The cross-linked samples were assessed with SDS-PAGE;about 30%-50%of the protein remains monomeric,whereas the remaining proteins correspond to the singly crosslinked form.
After trypsin(Promega)digestion,LC-MS/MS analysis was performed on an Easy-nLC 1000 UPLC(Thermo Fisher Scientifc)coupled with a Q Exactive Orbitrap mass spectrometer(Thermo Fisher Scientifc).The top ten most intense precursor ions from each full scan (resolution 70,000)were isolated for MS2 analysis.The pLink(Yang et al.2012)program was used to search a database containing the sequences of the proteins in question and the cross-linked peptides were identifed with the following criteria:false discovery rate smaller than 0.05 followed by an E-value cutoff of 10-3at the spectral level;at the peptide level,spectral count≥2 and the best E-value<10-8for each identifcation.The lower the E-value,the less likely the putative identifcation is a false discovery(Yang et al.2012).For each complex,the cross-linking reaction was repeated twice on different samples,which afforded almost identical cross-links.
To identify the intermolecular cross-links between two ubiquitin molecules,we mixed the15N-and14N-labeled(natural isotope abundance)ubiquitin at a 1:1 ratio.The14N-/14N-labeled and15N-/15N-labeled crosslinked peptide pairs were identifed using pLink(Yang et al.2012).Based on a strategy previously described (Taverner et al.2002;Petrotchenko et al.2014),we assigned cross-links between the15N and the14N-labeled peptides as intermolecular if the ratio in mass intensity in liquid chromatography of15N-/14N-labeled (or14N-/15N-labeled)cross-linked peptide relative to the corresponding14N-/14N-labeled (or15N-/15N-labeled)cross-linked peptide in the extracted ion chromatogram is>0.14.At this ratio,the intermolecular contribution is>25%.
Refnement of protein complex structures
The starting structures for the specifc complexes and for constituting proteins were retrieved from the PDB. The accession codes for trypsin,BPTI,and trypsin/BPTI complex are 4GUX,1JV8,and 2PTC,respectively.The accession codes for PP2Ac and PP2Ac/IGBP1 complex are 2NYL and 4IYP(Jiang et al.2013),respectively.Only the coordinates for the catalytic core domain were extracted from the PDB structure 2NYL.The coordinates for IGBP1 in the complex were obtained from the PDB structure 3QC1(free)and 4IYP(bound to PP2Ac). Since many residues in free IGBP1 structure are missing (residues V122-M144),the free structure was spliced with the bound structure,and the resulting structure was solvated in a cubic box containing the TIP3P water molecules with a 10 Å padding in all directions.The structure was subjected 10 ns MD simulation in Amber 14(Case et al.2012)to relax the conformation,to generate the initial coordinates for the unbound IGBP1. The accession code for the CDK9/Cyclin-T1 complex was 3BLH.The accession codes for EIN,HPr,and EIN/ HPr complexes were 1ZYM,1POH,and 3EZA(Garrett et al.1999),respectively.The PDB accession code forubiquitin monomer is 1UBQ(Vijay-Kumar et al.1987). The theoretical CXMS distance restraints for trypsin/ BPTI were calculated using Xwalk(Kahraman et al. 2011)with 24 Å cutoff.The intermolecular cross-links for PP2Ac/IGBP1 complex were taken from a previous study(Herzog et al.2012).In that report,the authors identifed seven cross-links,one of which involves IGBP1 Lys306;since the known structure for IGBP1 encompasses residues 1-221,this cross-link is not used for the structural refnement.
Structural refnement against the CXMS restraints was performed using Xplor-NIH (Schwieters et al. 2006).The refnement started from the coordinates for the free proteins.Each protein subunit was treated as a rigid body,and only CXMS and van der Waals repulsive terms between the subunits are considered.In the refnement,one subunit was fxed,and the other subunit was manipulated with a random rotation and translation,away from the fxed subunit.For each intermolecular cross-link,a square-well energy function was used to enforce the Cα-Cαdistance of the crosslinked lysine residues less than 24 and 20 Å for the BS3and BS2G cross-links,respectively(Lee 2009;Kahraman et al.2011).The upper limits of the distance restraints for cross-linking involving a protein N-terminus were 19 and 15 Å for the BS3and BS2G cross-linkers,respectively.The lengths correspond to a fully extended crosslinker and side chains of two cross-linked residues;no energy penalty was applied when the back-calculated Cα-Cαdistance was within the maximally allowed lengths.The penalty for a distance violation was defned as kΔ2,as the force constant k was gradually ramped from 1 to 30 kcal/(mol·Å2),as the bath temperature cooled from 3000 K to room temperature in the simulated annealing protocol.Upper limits for BS2G were used when intermolecular cross-links were observed with both BS2G and BS3;upper limits for BS3were used for intermolecular cross-links were observed with only BS3.In addition to the distance restraint derived from CXMS,the restraints also included covalent terms,and van der Waals repulsive energy term.For the ensemble refnement of ubiquitin homodimer,a C2non-crystallographic symmetry term was applied for each pair of interacting proteins.
For a protein complex,the structural refnement against CXMS restraints was frst performed with a single-conformer(N=1)representation for the complex.All the CXMS restraints could be satisfed for trypsin/BPTI and PP2Ac/IGBP1 complex.For EIN/HPr or ubiquitin/ubiquitin complexes,however,not all the cross-links could be accounted for.Thus we replicate the moving subunit to generate an N=2,3,4,or 5 ensemble to represent the complex,and different conformers in the ensemble can overlap.Ambiguous distance restraints were employed:each restraint was applied to the Cαatom of Lys(i)of the fxed subunit and to the Cαatom of Lys(j)of any conformer of the moving subunit,in which i and j are the residue numbers of cross-linked lysine residues in Table 1.We defned the CXMS energy to be related to inverse sixth power of the distance between the Cαatoms of two cross-linked residues,and to be averaged over all conformers in the ensemble.As a result,the CXMS term has a steep dependence on distance and is biased towards the conformer with the shortest Cα-Cαdistance,which can be satisfed providing that one of the conformers in the ensemble has shorter-than-maximum lysine Cα-Cαatom distance.The calculation was repeated 512 times starting from different random positions for each conformer of the moving subunit, and each calculation afforded a slightly different quaternary arrangement of the complex.Structures with no violations against CXMS restraints and no steric clashes were selected for further analysis.The fowchart for the ensemble refnement protocol against CXMS data was illustrated in Fig.S10.
The center-of-mass for one subunit with respect to the other subunit in the each CXMS model was calculated using an in-house Python script.The map projection with spherical coordinates was plotted using Gnuplot.The intermolecular NMR paramagnetic relaxation data were taken from previously published studies for EIN/HPr complex(Tang et al.2006;Fawzi et al. 2010)and for ubiquitin homodimer(Liu et al.2012), and ensemble refnement against the NMR data was performed as previously described.Reweighted atomic probability maps depicting the distribution of one subunit relative to another were calculated in Xplor-NIH (Schwieters et al.2006)and were plotted at respective thresholds(Schwieters and Clore 2002).Structural fgures were prepared with PyMOL(the PyMOL molecular graphics system).
Abbreviations
CXMS Chemical cross-linking of proteins coupled with mass spectrometry analysis
NMR Nuclear magnetic resonance
EM Electron microscopy
BS3Bis-sulfosuccinimidyl suberate
BS2G Bis-sulfosuccinimidyl glutarate
PDH Pimelic acid dihydrazide
BPTI Bovine pancreatic trypsin inhibitor
PP2Ac Phosphatase 2A catalytic subunit
IGBP1 Immunoglobulin binding protein 1
RMSD Root-mean-square deviation
Acknowledgments This work has been supported by grants from the Chinese Ministry of Science and Technology (2013CB910200),and the National Natural Science Foundation of China(31225007,31400735,31400644 and 21375010).The research of C.T.was supported in part by an International Early Career Scientist Grant from the Howard Hughes Medical Institute.
Compliance with Ethical Standards
Confict of Interest Zhou Gong,Yue-He Ding,Xu Dong,Na Liu,E. Erquan Zhang,Meng-Qiu Dong,and Chun Tang declare that they have no confict of interest.
Human and Animal Rights and Informed ConsentThis article does not contain any studies with human or animal subjects performed by the any of the authors.
Open Access This article is distributed under the terms of theCreative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricteduse, distribution, and reproduction in anymedium, provided you giveappropriate credit to the original author(s) and the source, provide alink to the Creative Commons license, and indicate if changes weremade.
Baumli S,Lolli G,Lowe ED,Troiani S,Rusconi L,Bullock AN, Debreczeni JE,Knapp S,Johnson LN(2008)The structure of P-TEFb(CDK9/cyclin T1),its complex with favopiridol and regulation by phosphorylation.EMBO J 27:1907-1918
Berg OG,Winter RB,Von Hippel PH(1981)Diffusion-driven mechanisms of protein translocation on nucleic acids.1. Models and theory.Biochemistry(Mosc)20:6929-6948
Case DA,Darden TA,Cheatham TEI,Simmerling CL,Wang J,Duke RE,Luo R,Walker RC,Zhang W,Merz KM,Roberts B,Hayik S, Roitberg A,Seabra G,Swails J,Goetz AW,Kolossva´ry I,Wong KF,Paesani F,Vanicek J,Wolf RM,Liu J,Wu X,Brozell SR, Steinbrecher T,Gohlke H,Cai Q,Ye X,Wang J,Hsieh MJ,Cui G, Roe DR,Mathews DH,Seetin MG,Salomon-Ferrer R,Sagui C, Babin V,Luchko T,Gusarov S,Kovalenko A,Kollman PA (2012)AMBER 12.University of California,San Francisco
Fawzi NL,Doucleff M,Suh JY,Clore GM(2010)Mechanistic details of a protein-protein association pathway revealed by paramagnetic relaxation enhancement titration measurements. Proc Natl Acad Sci USA 107:1379-1384
Gabdoulline RR,Wade RC(2002)Biomolecular diffusional association.Curr Opin Struct Biol 12:204-213
Garrett DS,Seok YJ,Peterkofsky A,Gronenborn AM,Clore GM (1999)Solution structure of the 40,000 Mr phosphoryl transfer complex between the N-terminal domain of enzyme I and HPr.Nat Struct Biol 6:166-173
Herzog F,Kahraman A,Boehringer D,Mak R,Bracher A, Walzthoeni T,Leitner A,Beck M,Hartl FU,Ban N,Malmstrom L,Aebersold R(2012)Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry.Science 337:1348-1352
Jiang L,Stanevich V,Satyshur KA,Kong M,Watkins GR,Wadzinski BE,Sengupta R,Xing Y(2013)Structural basis of protein phosphatase 2A stable latency.Nat Commun 4:1699
Jones S,Thornton JM (1996)Principles of protein-protein interactions.Proc Natl Acad Sci USA 93:13-20
Kahraman A,Malmstrom L,Aebersold R(2011)Xwalk:computing and visualizing distances in cross-linking experiments.Bioinformatics 27:2163-2164
Kahraman A,Herzog F,Leitner A,Rosenberger G,Aebersold R, Malmstrom L(2013)Cross-link guided molecular modeling with ROSETTA.PLoS One 8:e73411
Kalisman N,Adams CM,Levitt M(2012)Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking,mass spectrometry,and combinatorial homology modeling.Proc Natl Acad Sci USA 109:2884-2889
Kastritis PL,Moal IH,Hwang H,Weng Z,Bates PA,Bonvin AM, Janin J(2011)A structure-based benchmark for proteinprotein binding affnity.Protein Sci 20:482-491
LaskerK,ForsterF,BohnS,WalzthoeniT,VillaE,UnverdorbenP,Beck F,Aebersold R,Sali A,Baumeister W(2012)Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach.Proc Natl Acad Sci USA 109:1380-1387
Lee YJ(2009)Probability-based shotgun cross-linking sites analysis.J Am Soc Mass Spectrom 20:1896-1899
Leitner A,Joachimiak LA,Unverdorben P,Walzthoeni T,Frydman J, Forster F,Aebersold R(2014)Chemical cross-linking/mass spectrometry targeting acidic residues in proteins and protein complexes.Proc Natl Acad Sci USA 111:9455-9460
Liu Z,Zhang WP,Xing Q,Ren X,Liu M,Tang C(2012)Noncovalent dimerization of ubiquitin.Angew Chem Int Ed Engl 51: 469-472
Liu Z,Gong Z,Dong X,Tang C(2016)Transient protein-protein interactions visualized by solution NMR.Biochim Biophys Acta 1864(1):115-122
Lossl P,Kolbel K,Tanzler D,Nannemann D,Ihling CH,Keller MV, Schneider M,Zaucke F,Meiler J,Sinz A(2014)Analysis of nidogen-1/laminin gamma1 interaction by cross-linking, mass spectrometry,and computational modeling reveals multiple binding modes.PLoS One 9:e112886
Marquart M,Walter J,Deisenhofer J,Bode W,Huber R(1983)The geometry of the reactive site and of the peptide groups in trypsin,trypsinogen and its complexes with inhibitors.Acta Crystallogr B 39:480-490
Merkley ED,Rysavy S,Kahraman A,Hafen RP,Daggett V,Adkins JN (2014)Distance restraints from crosslinking mass spectrometry:mining a molecular dynamics simulation database to evaluate lysine-lysine distances.Protein Sci 23:747-759
Nilges M(1995)Calculation of protein structures with ambiguous distance restraints.Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities.J Mol Biol 245: 645-660
Nooren IM,Thornton JM(2003)Diversity of protein-protein interactions.EMBO J 22:3486-3492
Petrotchenko EV,Serpa JJ,Makepeace KA,Brodie NI,Borchers CH (2014)(14)N(15)N DXMSMS Match program for the automated analysis of LC/ESI-MS/MS crosslinking data from experiments using(15)N metabolically labeled proteins. J Proteomics 109:104-110
Plaschka C,Lariviere L,Wenzeck L,Seizl M,Hemann M,Tegunov D, Petrotchenko EV,Borchers CH,Baumeister W,Herzog F,Villa E,Cramer P(2015)Architecture of the RNA polymerase IIMediator core initiation complex.Nature 518:376-380
Politis A,Stengel F,Hall Z,Hernandez H,Leitner A,Walzthoeni T, Robinson CV,Aebersold R(2014)A mass spectrometry-based hybrid method for structural modeling of protein complexes. Nat Methods 11:403-406
Rappsilber J(2011)The beginning of a beautiful friendship:crosslinking/mass spectrometry and modelling of proteins and multi-protein complexes.J Struct Biol 173:530-540
Rinner O,Seebacher J,Walzthoeni T,Mueller LN,Beck M,Schmidt A,Mueller M,Aebersold R(2008)Identifcation of crosslinked peptides from large sequence databases.Nat Methods 5:315-318
Schilder J,Ubbink M(2013)Formation of transient protein complexes.Curr Opin Struct Biol 23:911-918
Schmidt C,Robinson CV(2014)Dynamic protein ligand interactions-insights from MS.FEBS J 281:1950-1964
Schreiber G,Fersht AR(1996)Rapid,electrostatically assisted association of proteins.Nat Struct Biol 3:427-431
Schwieters CD,Clore GM(2002)Reweighted atomic densities to represent ensembles of NMR structures.J Biomol NMR 23:221-225
Schwieters CD,Kuszewski JJ,Clore GM(2006)Using Xplor-NIH for NMR molecular structure determination.Prog Nucl Magn Reson Spectrosc 48:47-62
Suh JY,Tang C,Clore GM(2007)Role of electrostatic interactions in transient encounter complexes in protein-protein association investigated by paramagnetic relaxation enhancement. J Am Chem Soc 129:12954-12955
Tang C,Iwahara J,Clore GM(2006)Visualization of transient encounter complexes in protein-protein association.Nature 444:383-386
Tang C,Louis JM,Aniana A,Suh JY,Clore GM(2008)Visualizing transient events in amino-terminal autoprocessing of HIV-1 protease.Nature 455:U692-U693
Taverner T,Hall NE,O’Hair RA,Simpson RJ(2002)Characterization of an antagonist interleukin-6 dimer by stable isotope labeling,cross-linking,and mass spectrometry.J Biol Chem 277:46487-46492
Thalassinos K,Pandurangan AP,Xu M,Alber F,Topf M(2013) Conformational states of macromolecular assemblies explored by integrative structure calculation.Structure 21:1500-1508
The PyMOL molecular graphics system,Version 1.7.4 Schro¨dinger, LLC
Vijay-Kumar S,Bugg CE,Cook WJ(1987)Structure of ubiquitin refned at 1.8 A resolution.J Mol Biol 194:531-544
Vinogradova O,Qin J(2012)NMR as a unique tool in assessment and complex determination of weak protein-protein interactions.Top Curr Chem 326:35-45
Walzthoeni T,Leitner A,Stengel F,Aebersold R(2013)Mass spectrometry supported determination of protein complex structure.Curr Opin Struct Biol 23:252-260
Xing Q,Huang P,Yang J,Sun JQ,Gong Z,Dong X,Guo DC,Chen SM, Yang YH,Wang Y,Yang MH,Yi M,Ding YM,Liu ML,Zhang WP, Tang C(2014)Visualizing an ultra-weak protein-protein interaction in phosphorylation signaling.Angew Chem Int Ed Engl 53:11501-11505
Yang B,Wu YJ,Zhu M,Fan SB,Lin J,Zhang K,Li S,Chi H,Li YX, Chen HF,Luo SK,Ding YH,Wang LH,Hao Z,Xiu LY,Chen S,Ye K,He SM,Dong MQ(2012)Identifcation of cross-linked peptides from complex samples.Nat Methods 9:904-906
Zheng C,Yang L,Hoopmann MR,Eng JK,Tang X,Weisbrod CR, Bruce JE(2011)Cross-linking measurements of in vivo protein complex topologies.Mol Cell Proteomics 10:M110 006841
Zhou Gong and Yue-He Ding have contributed equally to this work.
Electronic supplementary material The online version of this article(
10.1007/s41048-015-0015-y)contains supplementary material,which is available to authorized users.
✉Correspondence:dongmengqiu@nibs.ac.cn(M.-Q.Dong),
tanglab@wipm.ac.cn(C.Tang)
杂志排行

Biophysics Reports的其它文章
- Implications for directionality of nanoscale forces in bacterial attachment
- E2-2,a novel immunohistochemical marker for both human and monkey plasmacytoid dendritic cells
- Radiolig and saturation binding for quantitative analysis of ligand-receptor interactions
- Pseudomonas sp.LZ-Q continuously degrades phenanthrene under hypersaline and hyperalkaline condition in a membrane bioreactor system
- Thermodynamics of GPCR activation