自动真空液相色谱装置的研制及其在五味子成分分离中的应用
2015-08-03朱靖博刘宝月单世波寇自农
朱靖博, 刘宝月, 单世波, 丁 燕,, 寇自农,3, 萧 伟
(1.大连工业大学食品学院,辽宁大连116034;2.大连工业大学植物资源化学与应用研究所,辽宁大连116034;3.大连工业大学实验仪器中心,辽宁大连116034;4.江苏康缘药业股份有限公司,江苏 连云港222001)
制备色谱是中药分离纯化过程的通用工具[1],已广泛应用于医药、精细化工等行业,但因自身存在的成本高、周期长、系统性及可重复性差等问题[2,3],往往花费大量时间、人力才能得到少量毫克级的纯品[4]。因此,在现有色谱分离理论及技术的基础上,研制自动的制备色谱装置以满足中药组分高效率分离制备需求,完成复杂药材组分到简单组分、单一化合物的系统分离,可能是解决中药研究中关键问题的有效方法[5,6]。
真空液相色谱(vacuum liquid chromatography,VLC)于 1979 年由 Targett等[7]命名,它利用柱后减压,使洗脱剂迅速通过固定相,从而很好地分离样品[8]。VLC实质上是柱色谱,与传统的柱色谱法相比,VLC具有设备简单、溶剂消耗少、回收率高、分离容量大、分离效果好,可以用薄层色谱(TLC)摸索分离条件,可以进行梯度洗脱等优点[9],目前已成功用于萜类、木脂素、生物碱等活性天然产物的分离[10,11]。然而,VLC 作为一种价廉高效、简便快捷的分离技术还存在诸多不足之处,如色谱柱填料装填不紧密导致柱床稳定性较差、理论塔板数低、色谱柱效不可控;每收集一个组分便要停止减压一次,使操作不连续[12];供液、真空洗脱等步骤以及梯度循环过程需要人工进行,消耗了大量的时间和人力。因此,保持柱床稳定,最大限度减少人工操作,实现VLC自动化分离,显著提升VLC分离的优越性是VLC研究的方向。目前尚未见有关于自动VLC的研制并应用于中药组分分离的报道。
本文研制了基于动态轴向压缩色谱柱的自动真空液相色谱(AUTO-VLC)装置,采用可编程逻辑控制器(PLC)实现了分离过程的自动控制及监测;采用该装置对五味子石油醚萃取物进行分离,取得了良好的结果。AUTO-VLC的研制及其应用对于中药成分自动和快速分离具有重要价值。
1 自动真空液相色谱的研制

图1 自动真空液相色谱系统结构及控制原理图Fig.1 Structure and control principle of automatic vacuum liquid chromatographic system
1.1 设计思想
采用动态轴向压缩色谱柱解决了传统VLC柱效不高、柱床不稳定和变化的问题,以S7-200 PLC对分离过程的不同比例流动相切换、不同规格色谱柱选择、分离时间设定及馏分收集、动态轴向压缩色谱柱的自动控制及监测,实现VLC对天然药物分离纯化的快速、有效、智能操控。
1.2 装置结构与控制
自动VLC系统结构及控制原理见图1。
AUTO-VLC由流动相储液系统、自动化控制系统、动态轴向压缩色谱分离柱系统和馏分收集系统等4部分组成,其中流动相储液系统有10个1 000 mL的玻璃或不锈钢容器,经支管连接至10进1出的气动控制阀,再经过1进3出的气动控制阀选择性进入3支长度均为280 mm,直径分别为80、100、150 mm的动态轴向压缩色谱柱上,以满足不同重量植物提取物分离的需求。不同色谱柱经过VLC分离的馏分经过3进1出和1进10出的气动控制阀进入由10个具有标准接口的玻璃馏分收集器,装置中的各种气动控制阀均采用自主设计的电磁开关控制气体驱动阀。电气自动化控制系统在面板上设有信号灯、人机界面(HMI)和控制按钮,在箱体内设有西门子S7-200 PLC、电磁阀、动态轴向压缩色谱柱的气驱液压系统。3个动态轴向压缩色谱柱采用液压油缸驱动柱中活塞完成柱床装填、压紧、稳定和卸填料。10个1 000 mL的具有标准接口的玻璃瓶与真空泵相连构成馏分收集系统。
自动控制系统中采用的西门子S7-200 PLC包括一个中央处理器(CPU)及5个扩展模块(控制按钮信息模块EM1、阀位置信息与控制模块EM2、阀通-断转换模块AM1、油缸控制模块AM2和信号控制模块AM3)。CPU负责执行逻辑运算和存储数据,S7-200 PLC的用户程序中包括阀位置逻辑控制、计数器、定时器、复杂数学运算以及与其他智能模块通讯等指令内容,从而使它能够监视输入状态,改变输出状态以达到控制目的。控制程序使用STEP 7-Micro/WIN编程软件作为用户开发、编辑和监控的应用程序。HMI输入和输出是系统的控制点,输入部分负责外部检测装置采集开关等状态信号,由CPU进行数学和逻辑运算,其结果由输出模件输出,通过驱动输出单元控制电磁阀气缸以及油缸运动方向,即可实现系统中每个气动控制阀按照分离要求进行通-断转换,完成分离过程的不同比例流动相切换、不同规格色谱柱选择、分离时间设定及馏分收集的自动控制及监测。EM1采集按钮信息,EM2采集阀组合的位置信息,AM1负责阀的切换,AM2控制动态轴向压缩色谱柱,AM3控制信号灯,如系统出现故障时蜂鸣器就会发出声光报警。本装置中的气动控制阀为多个截止阀组成的组合阀。切换指令为时间指令,可以根据实验目的人为设定。
1.3 操作流程
当分离样品时,选择相应色谱柱上样并在储液罐内注入不同比例的流动相,设定抽真空时间后选择自动运行方式,触摸HMI上启动按钮(或操作面板上的按钮),设备开始运行。阀组合按照设定时间自动切换,流动相连续流入到色谱柱内,分离后的流出液进入馏分收集器,实现自动分离。操作人员通过HMI可知当前的进展过程及状态。
2 自动真空液相色谱的分离应用
2.1 仪器、试剂与材料
仪器:自动真空液相色谱(自主研制);UltiMate 3000高效液相色谱仪(美国戴安公司);HGTZF-1三用紫外分析仪(上海精科实业有限公司);BS2245电子天平(北京赛多利斯仪器公司);R502B旋转蒸发仪(巩义市予华仪器有限责任公司)。
试剂:工业级乙酸乙酯、石油醚购自天津大茂化学试剂厂;分析级甲醇、乙酸乙酯、四氢呋喃、甲酸购自天津科密欧化学试剂有限公司;色谱级乙腈、甲醇购自美国Tedia公司。
材料:北五味子,产地为黑龙江。
2.2 实验方法
2.2.1 色谱条件
色谱柱:C18色谱柱(250 mm ×4.6 mm,5 μm;大连博迈科技发展有限公司);流动相:乙腈(A)和0.1% (v/v)甲酸水溶液(B);梯度洗脱程序:0 ~10 min,50%A~55%A;10~30 min,55%A ~60%A;30~52 min,60%A ~70%A;52~60 min,70%A~90%A。流速:1.0 mL/min;检测波长:250 nm;进样量:10 μL。
2.2.2 组分样品的分离
取1 kg五味子药材经乙醇80℃加热回流提取得到 SCB-crude,经石油醚萃取、浓缩、干燥得到SCB-ext。通过 TLC 法筛选填料、流动相[13],确定硅胶为分离介质。取2 000 g硅胶(300~400目)填充至150 mm×280 mm色谱柱管内,称取100 g SCB-ext与200 g硅胶干法混合样品,以石油醚/乙酸乙酯溶剂体系进行粗分离,不同比例流动相每份洗脱 1/6的柱体积(bed volume,BV),柱压为 -0.6 MPa,每一份流动相抽真空时间设为30 min,自动收集样品。利用TLC和HPLC检测后合并得到S1~S6共6份样品。
2.2.3 AUTO-VLC分离条件筛选与分离验证
取样品S5用乙酸乙酯溶解,配制成质量浓度为10 g/L的溶液并进行薄层制备,操作条件:使用200 mm×200 mm的薄层制备板,吸附剂厚度0.4~0.6 mm,条带点样,展开剂为石油醚-乙酸乙酯(70∶30,v/v),展开至距上沿2 cm处取出,在紫外灯下标注后刮取斑点I~R(见图2),刮取的斑点组分用乙酸乙酯-甲醇(1∶1,v/v)溶解并过实验室自制的固相萃取柱(直径1 cm,长度20 cm)去除硅胶,将乙酸乙酯-甲醇(1∶1,v/v)蒸干后加入四氢呋喃溶解,并进行HPLC分析。取薄层制备得到的样品I~R,以石油醚-乙酸乙酯为展开剂,调整其展开剂比例使化合物在薄层板上连续多次展开,紫外灯下观察并记录各点的比移值(Rf)和相邻斑点的分离度(R)[2,14],确定样品 S5 的梯度洗脱条件。

图2 样品S5及其经自动VLC分离后各样品的TLC谱图Fig.2 TLC chromatograms of sample S5 and all the samples from sample S5 after the separation by automatic vacuum liquid chromatography
应用上述得到的梯度洗脱条件,采用80 mm×280 mm真空制备色谱柱对2.2.2节中分离得到的样品S5进行分离,填料为700 g 200~300目硅胶,分离操作条件同2.2.2节(样品S5的质量即上样量)。利用TLC、HPLC逐一对样品进行检测,合并相同样品,得到各组分或化合物。
3 结果与讨论
3.1 五味子提取物的HPLC分析
根据文献[15]确定反相C18色谱柱为分析柱,乙腈(A)与0.1% (v/v)甲酸水溶液(B)为流动相,并在文献[15]的色谱条件基础上对流动相比例进行了调整,使五味子提取物达到了较好的分离(见图3)。
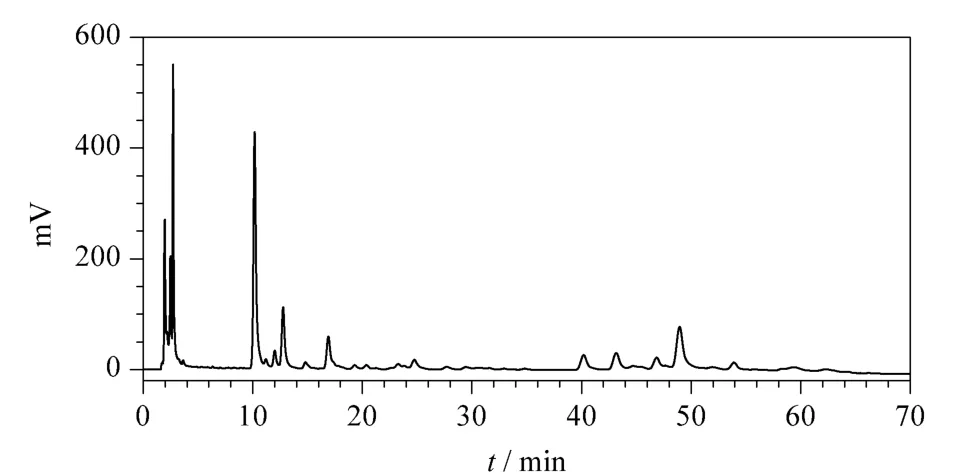
图3 五味子乙醇提取物的HPLC谱图Fig.3 HPLC chromatogram of Schisandra chinensis(Turcz)Baill.extract with ethanol
3.2 组分样品的分离
SCB-crude经石油醚萃取浓缩干燥得到SCB-ext。通过TLC筛选,确定分离填料为硅胶,洗脱剂为石油醚和乙酸乙酯,两者的体积比例按极性由小到大依次为100∶0、95∶5、90∶10、85∶15、80∶20、75∶25、70∶30、65∶35、60∶40、55∶45、50∶50、45∶55、40∶60、35∶65、30∶70、25∶75、20∶80、15∶85、10∶90、5∶95、0∶100。100 g SCB-ext经自动 VLC分离及 TLC、HPLC检测后合并相同组分,共得到6个样品S1~S6(见图4),其质量分别为1.12、64.86、14.24、1.7、5.71、11.5 g。可知自动VLC对五味子萃取物复杂组分能够起到快速集中分段的作用,同时证明PLC对本装置的自动控制及监测是有效的。
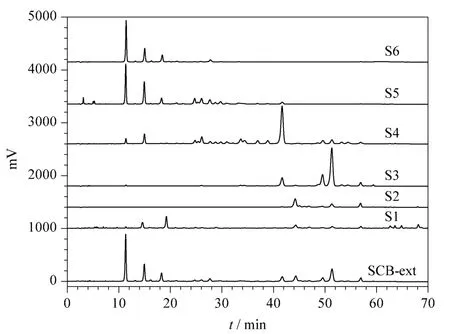
图 4 SCB-ext及6个样品(S1~S6)的HPLC谱图Fig.4 HPLC chromatograms of SCB-ext and the six samples(S1-S6)
3.3 AUTO-VLC分离条件筛选与分离验证
VLC在进行溶剂洗脱时与制备薄层色谱的多次展开极为相似[7],故选定 TLC多次展开作为AUTO-VLC分离条件的筛选方法。一般认为被分离物质对分离度较大且化合物Rf值为0.3[16]时的洗脱强度适合于色谱柱分离,选择石油醚与乙酸乙酯的体积比分别为 85∶15、83∶17、81∶19、77∶23、72∶28、65∶35、55∶45 对样品 S5 进行连续多次展开,得到展开次数与Rf的线性函数斜率(k)的变化趋势。由图5可知,当洗脱溶剂比例不断变化时,在0<Rf<0.45范围内,k呈上升趋势;在0.45<Rf<0.6时,k开始下降;0.6<Rf<0.8时,k下降趋势减慢;Rf>0.8后,k下降趋势加快。在此基础上确定了将待分离全部目标化合物展开至Rf为0~0.45的初始展开剂为起始洗脱溶剂比例。
为有效实现样品中各个化合物的分离,采用同一溶剂比例多次洗脱,洗脱次数n由TLC上Rf最小的目标化合物(即移动速率最慢的化合物)在此条件下的线性函数依照计算公式n≈ΔRf/k确定(假定化合物Rf与展开次数呈线性相关;ΔRf为化合物多次展开的Rf值之差)。在Rf>0.45时,则增加极性溶剂比例,且增加趋势不断加大。每次洗脱体积为1/4 BV。以样品S5为AUTO-VLC分离的样品,以不同体积比的石油醚与乙酸乙酯为洗脱剂,最终确定了其洗脱程序,即起始洗脱剂的比例选择85∶15,洗脱次数由公式n≈ΔRf/k计算为12次。斜率k值依据不同比例多次展开时其变化趋势及TLC多次展开筛选条件时不同比例洗脱剂下的k值进行估算,从而计算梯度洗脱的次数。当0.45 <Rf<0.6 时,选择83∶17 的洗脱剂洗脱2次,81∶19的洗脱剂洗脱2次;当0.6<Rf<0.8 时,选择77∶23 的洗脱剂洗脱3 次,72∶28 的洗脱剂洗脱4次;当0.8<Rf<1时,选择65∶35和55∶45的洗脱剂分别洗脱6次和5次(见图5)。
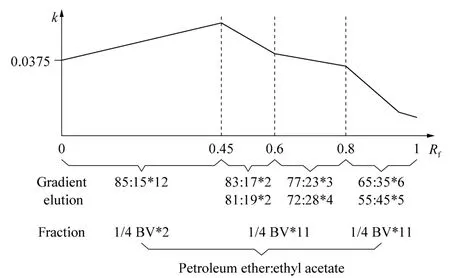
图5 斜率k的变化趋势及石油醚-乙酸乙酯梯度洗脱的比例与体积分数Fig.5 Variation trend of the slope k and the ratio and volume fractions of gradient elution consisting of petroleum ether-ethyl acetate
样品S5经AUTO-VLC分离得到34个组分(V1~V34),耗时约17 h。合并相似组分得到17个样品,极性由小到大依次命名为1~17。在254 nm波长下观察 TLC 分离结果(见图2),化合物 I、J、L、Q、R均得到了分离。HPLC检测结果见图6,其中化合物I、L、Q、R的纯度大于85%,且 Q、R 的纯度大于90%。可见AUTO-VLC分离快速且效果理想。

图6 样品S5中分离得到的5个化合物的HPLC谱图Fig.6 HPLC chromatograms of the five compounds from sample S5
4 结论
本文研制了AUTO-VLC装置并用于五味子石油醚萃取物的分离。该装置由自主设计的流动相储备系统、10通分流切换阀、3通切换阀、3个不同规格动态轴向压缩色谱柱、10通馏分收集阀和馏分收集器组成,采用S7-200 PLC实现了分离过程的不同比例流动相切换、不同规格色谱柱选择、分离时间设定及馏分收集的自动控制及监测。建立了多次TLC展开筛选VLC分离条件的方法并进行了分离验证。AUTO-VLC装置的研制实现了对复杂样品的快速、有效、自动化分离,具有良好的应用价值。
[1] McChesney J D,Rodenburg D L.Curr Opin Biotechnol,2014,25:111
[2] Kou Z N,Zhu J B,Wang X N,et al.China Journal of Traditional Chinese Medicine and Pharmacy(寇自农,朱靖博,王西宁,等.中华中医药杂志),2014,29(12):4014
[3] Liu N.Journal of Mathematical Medicine(刘宁.数理医药学杂志),2014,27(5):574
[4] Lei Q B.[MS Dissertation].Guangdong:Guangdong University of Technology(雷桥兵.[硕士学位论文].广东:广东工业大学),2010
[5] Wang T F.[MS Dissertation].Suzhou:Suzhou University(王天锋.[硕士学位论文].苏州:苏州大学),2012
[6] Feng J T,Xu Qing,Xue X Y,et al.World Science and Technology/Modernization of Traditional Chinese Medicine and Materia Medica(丰加涛,徐青,薛兴亚,等.世界科学技术-中医药现代化),2006,8(3):95
[7] Targett N M,Kilocyne J P,Green B.J Org Chem,1979,44:4962
[8] Hu K,Dong A J,Yao X S.Journal of Shenyang Pharmaceutical University(胡柯,董爱军,姚新生.沈阳药科大学学报),1995,38(7):146
[9] Xu R S.Natural Products Chemistry.Beijing:Science Press(徐任生.天然产物化学.北京:科学出版社),1993
[10] Chen Y G,Zhang Y,Feng L P.Yunnan Chemical Technology(陈业高,张燕,冯丽萍.云南化工),2000,27(5):19
[11] Sun W J,Men Y X,Zhang L,et al.Chinese Journal of Pharmaceutical Analysis(孙文基,门瑛璇,张利,等.药物分析杂志),1997,17(4):276
[12] Zheng G C.Chinese Journal of Organic Chemistry (郑国墀.有机化学),1989,9(3):286
[13] Wang X N,Zhu J B,Ding Y,et al.Northern Horticulture(王西宁,朱靖博,丁燕,等.北方园艺),2013(14):107
[14] Zhu J B,Han C M,Ding Y,et al.Natural Product Research and Development(朱靖博,韩聪敏,丁燕,等.天然产物研究与开发),2014,26(1):87
[15] Yin F Z,Yin W,Zhang X,et al.Acta Chromatogr,2010,22(4):609
[16] Nyiredy S,Dallenbach-Toelke K,Zogg G C,et al.J Chromatogr A,1990,499:453
