助剂对煤基合成气甲烷化反应用镍基催化剂的促进作用
2015-07-25张旭王子宗陈建峰
张旭,王子宗,陈建峰
(1 中国石化工程建设有限公司,北京 100101;2 北京化工大学化学工程学院,北京 100029)
甲烷化反应是一氧化碳、二氧化碳加氢生成甲烷的过程,同时也是以煤、生物质为原料经气化制取替代天然气(substitute natural gas,SNG)的重要途径[1]。近年来,环境法规日益严格,天然气需求量不断攀升。天然气燃烧后产生的温室气体只有煤炭的1/2、石油的2/3,因此,将煤、生物质转化得到的合成气或裂解气进行甲烷化反应技术受到越来越多的关注[2]。
煤通过气化制得合成气,我国炼焦行业还伴生焦炉煤气,合成气、焦炉煤气经过净化、变换及甲烷化等工艺过程后可以制得合格的替代天然气。煤制替代天然气的能量转化率高达56%,远高于煤发电、煤制油和煤制甲醇等,其热值可达到8500kcal(1kcal=4.186kJ)[3],不仅实现了对煤的高效利用,同时也为炼焦行业的焦炉煤气利用和补充我国天然气缺口提供了一条切实可行的途径。
法国科学家Sabatier 和Senderens 于1902年首次发现氢气和一氧化碳能够在镍基及其他金属(Rh、Ru、Pt、Fe、Co 等)基催化剂上合成甲烷。20 世纪70年代,受世界能源危机的影响,Topsøe、Davy、BASF 等企业加快了甲烷化技术的研究步伐,并率先在美国建成Great Plains 煤制天然气厂,从1984年建成投产至今已稳定运行三十年[4]。国内开展甲烷化技术开发起步较晚,20 世纪80~90年代,大连化学物理研究所在低热值煤气甲烷化制取中热值城市煤气方面进行了大量的研究工作,并相继应用到10 个工厂。国内在建和拟建的SNG 项目都采用引进的成套工艺技术,为此需要花费大量的技术使用费。目前,国内科研机构正在进行具有自主知识产权的甲烷化工艺技术开发,但是在甲烷化催化剂和配套工艺方面还未实现工业应用。
合成气中CO、CO2和H2除了发生甲烷合成反应外[式(1)、式(2)],还伴随有水煤气变换、歧化反应、甲烷裂解、一氧化碳和二氧化碳还原等副反应。从热力学角度看,CO 和CO2都可以进行甲烷化反应,均是强放热反应,并且反应过程伴随着析碳反应[5]。因此,研制煤制SNG 甲烷化催化剂必须具有较高的宽温活性、高甲烷选择性、抗积炭、抗高温烧结以及抗硫中毒能力。目前,甲烷化催化剂生产中使用的载体主要是SiO2[6]、γ-Al2O3[7-9]和ZrO2[10-11]等,活性组分以金属Ni[12]、Ru[13]等为主。Ni 由于其催化活性好、甲烷选择性高、储量丰富、价格低等特点,被广泛运用于甲烷化反应。
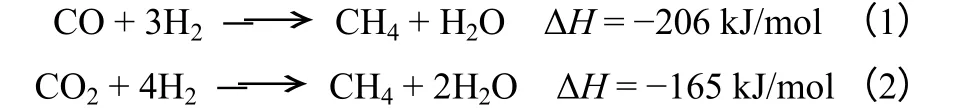
贵金属[14]、碱土金属[15]、稀土金属[16-17]以及过渡金属[18-19]是镍基催化剂常用的助剂。研究发现,助剂改性的镍基催化剂具有反应活性高、使用寿命长以及甲烷选择性高等优点[20-22]。甲烷化反应活性、选择性与活性镍物种结构有直接关系。甲烷化研究进展以往的报道主要侧重于甲烷化催化剂分 类[23-24]、工艺条件[25]以及反应机理[2,26]等方面,对于助剂改性的镍基分散度、还原度以及双金属协同效应,镍基催化剂结构稳定性及其对合成气甲烷化反应速率和产物选择性影响等方面等未见报道。本文结合最新的一些研究进展和专利技术,全面介绍了助剂对合成气甲烷化用镍基催化剂的改性作用,以期对开发具有自主知识产权的甲烷化催化剂起到积极作用。
1 助剂对甲烷化用镍基催化剂的 影响
添加助剂通过影响活性镍物种的晶粒尺寸、分散度、还原度、催化剂表面酸碱性以及热稳定性等,达到对甲烷化反应速率和产物分布调控的目的。根据助剂作用不同,分为结构助剂、电子助剂和晶格缺陷助剂三大类。助剂一般是通过载体浸渍镍金属盐和助剂金属盐的混合溶液引入。目前,对甲烷合成用镍基催化剂活性组分镍的纳米晶粒尺寸效应还尚存争议,但大量文献实验结果表明:甲烷化反应活性与催化剂表面的活性中心密切相关,活性中心数越多,催化剂活性也越高。镍基催化剂的还原 度、分散度与催化剂表面的镍活性中心数多少紧密相关[27]。
1.1 助剂对镍基催化剂还原性能的影响
氧化态的镍对甲烷化反应没有催化活性,在进行反应前需先将氧化态的镍在氢气气氛中进行还原,即NiO→Ni0。添加少量的Mg、Mn、Zr、Ce等助剂能显著改善镍基催化剂的还原性能,降低活性物种与载体之间的相互作用力,使得活性镍物种的还原能力提高。杨霞等[15]发现,MgO 助剂的添加可以明显提高Ni/γ-Al2O3催化剂的还原度。未添加MgO 助剂的Ni/Al2O3催化剂还原度为59.0%,而添加质量分数5%Mg 改性的Ni-Mg/Al2O3催化剂还原度为74.9%,可见Mg 改性提高了Ni/Al2O3催化剂还原度近16%。最近,HR-TEM 和H2-TPR 等表征技术也表明Zr、Co、Ce、Zn、La 的添加能够减小Ni 的晶粒尺寸,降低催化剂还原温度,并以La 助剂的效果最为明显[28]。研究发现,La、Ce 等稀土氧化物助剂是较好的电子给予体,当Ni 与载体Al2O3相作用后,电子由镍金属向载体转移,使得镍处于缺电子状态,La、Ce 等稀土氧化物与镍相作用后,电子向镍转移,缓解镍表面的缺电子状态有利于吸附活化CO 分子[29-30]。
添加助剂在一定程度上能够降低镍基催化剂的还原温度,增加催化剂表面的活性中心数,提高催化剂的催化性能。图1 给出的是Mn 改性前后Ni/γ-Al2O3催化剂的TPR 图谱。从图1 中可以看出,Mn 助剂的添加导致Ni-Mn/γ-Al2O3催化剂还原峰向低温方向迁移。具体而言,460℃的H2还原峰归属于与Al2O3载体间具有弱相互作用的NiO 还原,而560℃的还原峰则归属于与载体之间有强相互作用的NiO还原;Ni-Mn/γ-Al2O3催化剂由于Mn 的添加,使得还原峰峰形更加宽化,并向低温方向移动,出现了530℃的主耗氢峰和410℃的肩峰,主峰温度向低温移动了30℃。Mn 助剂的添加阻碍了Ni2+离子进入Al2O3表面的四面体和八面体空穴,降低了活性金属和载体之间的作用力,从而降低了还原温度。另外,结合H2-TPSR 分析结果可知,Mn 的加入并未改变催化剂表面活性中心的类型,也未出现新的活性位,但由于活性金属分散度和还原温度的降低,使得中等强度活性中心数量增多,是提高甲烷化催化剂活性的根本原因。
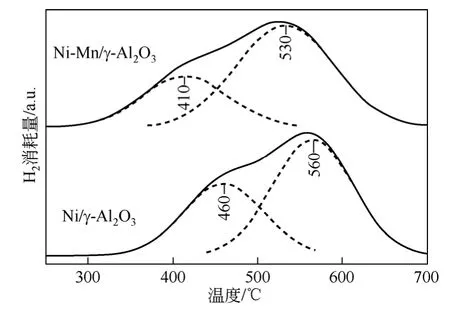
图1 Mn 改性的Ni/γ-Al2O3 催化剂TPR 图谱 [18]
镍晶相的形成经历了晶核的形成与生长两个阶段。ⅣB、ⅤB、ⅥB 和ⅦB 族过渡金属由于拥有未充满的d 电子轨道,起到了“电子转移站”的作用,所以添加少量的助剂有利于H2在催化剂表面上的吸附、活化和解离。相对而言,过渡金属氧化物能够在相对较低的温度下还原,形成的还原态金属进一步活化H2分子,将活化的H2以溢流方式转移至NiO 界面,促进了NiO 的还原,从而降低了还原温度,其作用过程见图2。从实验研究结果可知,过渡金属助剂对镍物种还原性能的影响可能与形成双金属颗粒或合金有关,产生了双金属协同作用。此外,助剂对镍基催化剂还原性能的影响在一定程度上还与载体性质有关。
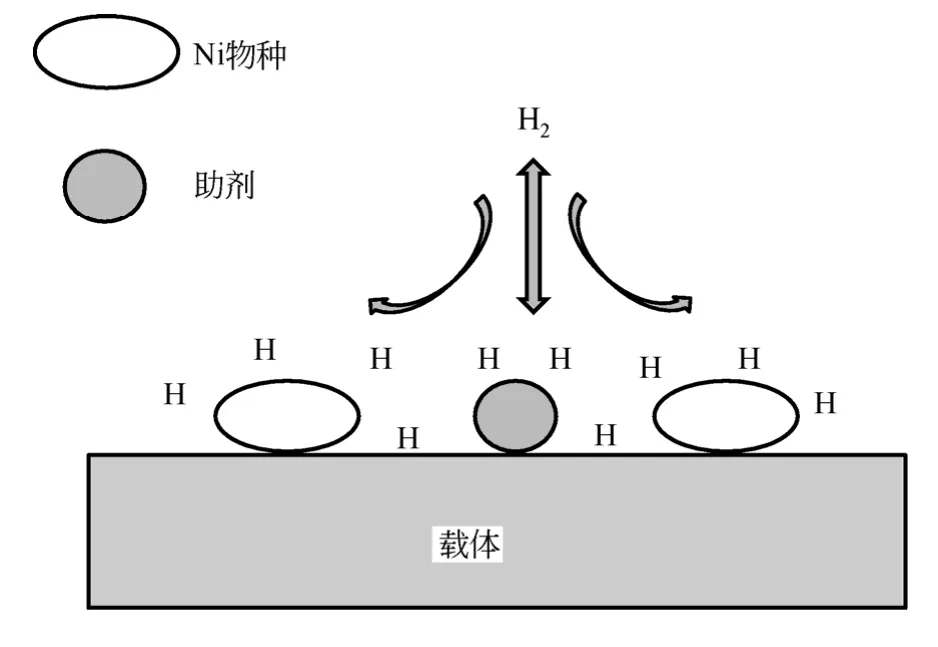
图2 过渡金属改性的镍基催化剂氢溢流示意图
1.2 助剂对镍基催化剂分散度的影响
活性镍物种在催化剂表面的分散度越高,其催化性能也越好。助剂的添加能够改善活性镍物种的分散程度,尤其是对于镍物种与载体作用力较弱的分散程度越明显。Mg 改性的 Ni/Al2O3催化剂镍分散度从11.8%提高到20.5%[15]。Wang 等[19]和Cai等[31]发现,ZrO2助剂可以提高Ni 物种在Al2O3和SiO2表面上的分散度,并且细化晶粒尺寸。在Ni/Al2O3催化剂中,添加一定量的CeO2或La2O3助剂有利于镍物种的分散。Liu 等[16]研究表明,Ni-2%CeO2/Al2O3具有最高的催化活性,随着CeO2质量分数的增加,催化剂表面活性位被占据,降低了催化剂的催化活性。La2O3能够很好地分散活性金属镍,使得活性组分晶粒度减小[32]。
助剂的添加能够影响催化剂镍颗粒的平均尺寸大小,可以通过SEM 定性给出活性物种在载体表面的分布情况,而XRD、TEM 表征技术可以获得定量的晶粒尺寸。图3(a)、图3(b)是Ce 改性的Ni/SiO2催化剂TEM 图,图3(c)、图3(d)是对应的晶粒大小分布图。Ce 改性的Ni/SiO2催化剂NiO 晶粒尺寸由未改性时的10~13nm 下降到5~8nm,与XRD 表征结果相吻合[6]。李凤梅等[33]采用等体积浸渍法制备了质量分数2% Ce 促进的7%Ni/SiO2样品,NiO 颗粒尺寸从9.2nm 下降到7.5nm。
还原态Ni-Co/Al2O3催化剂的磁场曲线如图4所示[7]。图中磁滞线的矫顽力 Hc=0,说明Ni-Co/Al2O3催化剂具有超顺磁性,同时也意味着NiCo 晶粒尺寸小于其具有单磁范围的临界尺寸(15nm)[34],可见Ni 物种具有较好的分散度。
添加合适的助剂能够提高镍基催化剂的分散度和还原度,如Fe、Mg、Ca、Ce 或 Zr 助剂等。Ce和Zr 较其他助剂在低温区具有明显的促进作用,并且Zr 促进的Ni 基催化剂在低温区对甲烷化反应具有较好的催化性能。Zhang 等[8]研究发现,采用共浸渍法制备Zr 促进的Ni/γ-Al2O3催化剂,在空速为20L/(g·h)的浆态床反应器中具有较好的低温活性。Zr 能够调节Ni 与载体γ-Al2O3之间的相互作用力,提高了Ni 晶粒活性中心数。在相同Ni 负载量的情况下,高浓度的Ni 晶粒活性中心能够形成大量的小颗粒镍物种,所以提高了镍催化剂的分散度。
1.3 双金属纳米颗粒协同效应
添加过渡金属、稀土金属等与活性金属镍相互作用,通过焙烧、氧化还原等方式形成双金属纳米颗粒或合金。Yu 等[35]采用XRD 表征发现,在Ni-Co/SiC 催化剂上有Ni-Co 合金存在。双金属纳米颗粒或合金在还原气体的作用下可以在较低的温 度下分别还原成金属镍和还原态金属。Tian 等[36]采用浸渍法制备了不同Fe 含量促进的Ni/γ-Al2O3催化剂。通过H2-TPR 表征可知,在一定Fe/Ni 比例范围内Ni-Fe/γ-Al2O3催化剂只有一个耗氢峰,这也是双金属催化剂形成合金的一个证据[37]。王宁、Kustov 等[38-39]研究结果表明,制备的Ni-Fe/γ-Al2O3双金属催化剂在还原后会形成Ni-Fe 合金,增加了活性镍物种数目,对CO、H2的吸附活化能力增强。
图3 13Ni/Si 和7Ni-2Ce/Si 催化剂TEM 图、晶粒尺寸 分布[6]

图4 还原态Ni-Co/Al2O3 催化剂的磁场曲线[7]
原位表征技术有助于认识活性中心和反应机理。镍基催化剂通常使用反应物CO 分子作为探针分子,用以表征载体表面不同的镍物种。催化剂表面存在着不同类型的镍物种,镍金属物种,氧化镍物种以及金属镍与助剂、载体发生相互作用的无定形镍物种、双金属合金等。镍基催化剂表面吸附的CO 活性顺序为:桥式吸附>线式吸附>孪式吸附。武瑞芳等[40]采用DRIFTS 分析ZrO2促进的Ni/SiO2催化剂时发现,在有氢气存在的情况下,ZrO2的添加大大提高了催化剂表面桥式羰基氢化物物种的数目。桥式吸附物种越多,C—O 键断键的概率越大,表面生成的活性碳物种也越多,活性碳物种是生成CH4的直接来源,这也直接证明了助剂的添加能够提高CH4的选择性。
Huang 等[11]采用H2-TPD 考察了Yb 改性的Ni/ZrO2催化剂对氢的吸附性能。研究结果发现,Yb 助剂的添加使得催化剂产生了两个较明显的H2脱附峰,脱附峰面积高于未添加助剂样品,323~473K 的低温区脱附峰对应于分子吸附态H2,473~973K 的高温区脱附峰则对应于解离吸附的氢。添加Yb 助剂增加了催化剂的吸氢量,提高了镍物种的还原度。高温区解离吸附氢对CO 甲烷化的反应性能直接相关,这从反应活性评价数据得到印证。另外,Yb 助剂的添加还对CO 分子吸附活化具有促进作用。添加助剂的镍基催化剂H2-TPD 高温区脱附峰温在750~850K 之间,该温度区间有部分溢流到载体上的氢脱附,也正是合成气甲烷化反应的反应温度区间,产生的溢流氢会加速甲烷化反应进程,这也是提高目标产物甲烷选择性的原因之一。
程序升温表面反应(TPSR)是在程序升温过程中研究表面化学反应与产物脱附过程的表征技术。通过吸附在催化剂表面的CO 在程序升温的条件下加氢,可以表征活性中心的性质。CO 加氢生成的CH4分子在催化剂表面的吸附作用力较弱,因此可以通过CH4脱除的温度高低间接反映出催化剂活性大小。稀土金属的添加增强了镍物种对CO 的中低温解离能力,所以提高了吸附态CO 的加氢活性[17]。
对于助剂改性的镍基催化剂,金属合金是通过镍金属与助剂金属直接成键形成的。近年来,XANES/EXAFS 表征技术运用于金属助剂改性的镍基催化剂协同效应研究。Wang 等[41]利用EXAFS 技术分析了Pt-Ni 分别在γ-Al2O3、SiO2、TiO2、CeO2和ZrO2载体上的双金属协同作用。Pt 的LⅢ-边EXAFS 分析发现Pt-Ni 形成了双金属颗粒[42],Pt-Ni键长在2.50~2.58Å(1Å=0.1nm),介于Ni-Ni 键(2.49Å)和Pt-Pt 键(2.77Å)之间。陈豫等[43]同样利用EXAFS 分析了La、Na、Mg、Ba 改性的Ni/γ-Al2O3催化剂中的双金属作用。
2 助剂对镍基催化剂抗失活行为的影响
反应活性高低、使用寿命长短是评价工业催化剂性能好坏的关键指标。镍基催化剂失活的主要影响因素有表面积炭、烧结和硫中毒等,催化剂失活导致甲烷化过程成本难以降低。通过添加助剂、载体以及改变催化剂生产工艺,可以在一定程度上改善催化剂的抗积炭、热稳定性和抗中毒性能。
甲烷化反应是一个强烈的放热反应,反应过程中常伴随有副反应的发生。表1 给出了甲烷化过程中可能发生的化学反应。其中,反应式(6)~式(8)会造成催化剂床层和反应器积炭。反应放出的热量很容易破坏催化剂结构并造成活性物种镍的团聚,必须及时移除反应热,防止催化剂过热失活,每1%CO 转化为甲烷会引起床层63℃绝热温升。
Guo[12]、田大勇[44]等研究表明,ZrO2的添加能够有效抑制镍基催化剂的积炭。此外,田大勇等还发现MoO3助剂对催化剂的活性和抗硫性均有明显的改善。MoO3促进Ni/MCM-41 催化剂失活的原因在于催化剂结构烧结而非积炭[45]。Liu 等[9]在研究Ni-Mg/Al2O3催化剂用于甲烷化反应时,在催化剂表面形成两种不同类型碳。在甲烷化反应过程中,活性镍物种表面容易积炭。稀土金属氧化物Ce 能够提供更多的氧空位,抑制催化剂表面碳的形 成[46];La 的引入会增加Ni 基催化剂表面储氢能力,有效地预防了催化剂积炭[47],同时还可以提高催化剂的热稳定性[48]。

表1 甲烷化过程中可能发生的反应[5]
碱土金属由于有较好的传热性能,通常被作为助剂添加到强放热催化剂中。MgO 与NiO 两者都具有NaCl 结构的立方晶格,且离子半径相近[24]。MgO 与Al2O3通过高温焙烧可以形成具有镁铝尖晶石MgAl2O4的稳定结构。镁铝尖晶石结构与活性组分不发生反应,MgO 的加入调变了Al2O3表面酸碱性,使得催化剂载体表面呈中性或弱碱性,抑制了积炭的发生,延长了催化剂寿命。杨霞等[15]和郭雄等[49]均证实,MgO 改性的Ni/Al2O3催化剂形成了具有稳定结构的镁铝尖晶石结构,催化剂表现出了较高的力学强度、抗积炭性能、低温活性和热稳 定性。
镍基催化剂容易受到硫中毒影响而降低催化活性,所以在工业甲烷化合成段之前添加了合成气的水解和低温甲醇洗等净化装置,将合成气中总硫含量降低到50×10-9以下。钼的添加可显著地改善镍基催化剂的抗硫性,主要是基于钼助剂的结构效应和电子效应[50]。苗茵等[51]从Ni-Mo/γ-Al2O3催化剂的CO-TPD 和XRD 分析得到,添加钼后催化剂的吸附中心类型和数目发生了变化,CO 吸附量明显增加,衍射峰明显宽化,镍晶粒变细,充分说明钼提高了镍物种的分散度。另外,结合催化剂的XPS可知,当添加钼助剂后Ni2P3/2的结合能发生了负的化学位移,使得金属Ni 上略带负电荷,这提高了Ni 表面吸附CO 的2π*反键轨道的电子反馈能力,增强了CO 与Ni 的相互作用,削弱了C—O 键,使之易于断裂,有利于甲烷化反应进行。同时,由于钼的加入,原料气中的硫首先与钼反应生成二硫化钼,二硫化钼的生成阻止了硫与活性物种镍的作用,延缓了催化剂硫中毒。可见正是钼助剂的结构效应和电子效应双重作用提高了镍基催化剂的甲烷选择性和抗硫性。目前对抗硫催化剂研究较多,但是钼基抗硫催化剂的反应活性不高,需要加压操作,对设备提出了更高的要求,限制了工业应用。为此,可尝试对其进行稀土或过渡金属掺杂改性。
3 助剂对甲烷化反应速率和产物分布的影响
甲烷化反应产物的分布(甲烷选择性)是研究甲烷化反应机理的出发点。甲烷化反应是一个快速、体积缩小的强放热反应。从热力学角度看,低温高压有利于反应的进行;从动力学角度看,高温高压有利于反应的进行。目前CO 甲烷化反应机理的分歧主要是:①CO 是直接解离还是氢助解离;②反应的速率控制步骤是CO 解离还是表面碳加氢。CO2甲烷化反应由早期生成“CO”中间体的反应机理逐渐被“碳酸根”和“甲酸根”中间体反应机理所 代替。
研究表明,CO 存在条件下CO2甲烷化反应速率较小,主要发生CO 甲烷化反应。CO 的存在对CO2甲烷化反应有抑制作用,而CO2的存在对CO甲烷化反应无明显阻碍作用[52]。CO 作为极性分子易于吸附于镍基活性位,占据了部分CO2甲烷化的活性中心位,优先发生甲烷化反应,随着CO 浓度下降到一定程度,CO 和CO2之间会形成竞争甲烷化反应[53]。同一催化剂CO2甲烷化反应速率高于CO,说明了CO2甲烷化反应生成CH4不需要经过逆水煤气变换反应,而是形成含氧酸根中间体,最后含氧酸根进一步加氢生成CH4[54]。
甲烷化镍基催化剂的反应活性和产物分布与助剂的添加密切相关。由于不同类型的助剂与活性镍物种的作用机制不同,导致了不同催化性能。催化剂催化活性大小主要体现在对一氧化碳的解离能高低和主要中间体的稳定性强弱[55]。
不同过渡金属助剂改性的Ni/ZrO2催化活性呈现 出 如 下 顺 序:Ni-Mn/ZrO2>Ni-Cr/ZrO2>Ni-Co/ ZrO2>Ni-Fe/ZrO2>Ni-Cu/ZrO2,由于Cu 具有充满电子的d 轨道,影响了金属Ni 周围的电子分布,降低了活性镍物种对反应物分子的吸附活化作用,所以其催化活性最低[10]。崔晓曦等[28]对比了Zr、Co、Ce、Zn 和La 助剂对Ni/γ-Al2O3催化剂的甲烷化催化性能,掺杂Zr、Co、Ce、Zn 和La 助剂提高了催化剂的甲烷化性能,其中以 12%Ni-8%La/γ-Al2O3催化剂的效果最明显,CO 转化率、CH4选择性和时空收率分别达到了96.3%、87.1%和179.6g/(kg·h)。从表2 中也可以看出,甲烷化活性大小顺序为Ni-La>Ni-Zn>Ni-Ce>Ni-Co>Ni-Zr>Ni-Ni,他们将这种现象归结于不同助剂的作用机制不同。La 助剂有利于调控活性镍物种与载体之间的作用力,Zn、Zr等助剂有利于镍物种的分散,Ce 则能提高催化剂的还原性能。稀土金属助剂La、Ce 等改性的Ni 基催化剂,其反应活性、热稳定、抗积炭能力、活性镍物种分散度都得到了显著地提高,这主要与改性后催化剂表面形成更多的活性中心有关。根本原因在于稀土金属助剂能调变金属镍的电子状态,改善镍原子的缺电子状态。另外,稀土金属助剂比碱土金属助剂具有更明显的改性作用,同时添加两种助剂效果更佳。因此,在甲烷化催化剂添加稀土元素助剂将成为提高催化剂活性的一个有效途径。
贵金属Ru 也是一种优异的甲烷化反应催化剂活性组分。它的优点主要是反应温度低,尤其是反应的起活温度低,同时甲烷选择性高。虽然其储量少、价格昂贵限制了在工业催化剂生产中的大量应用,但是经常在工业催化剂中添加少量的Ru,用来改善催化剂的催化活性和提高目标产物的收率。Kimura 等[53]报道,1% Ru/NiAlxOy(500)具有很高的CO 加氢甲烷化活性和CH4选择性,主要原因是:①贵金属Ru 能够产生H2溢流现象,促进了活性组分的还原,提高了对H2的活化能力;②抑制了原料气中CO2在活性位解离为CO 过程的发生。Tada 等[13]还发现,在Ni-Ru/TiO2催化剂中,随着Ru 含量的增加,逆水煤气变换反应的速率也之增加,0.2% Ru 的添加量较为合适;Ru-Ni/TiO2催化剂进行5500h 的稳定性试验仍然具有较高的活性,并且保持较宽的温度窗口[56]。另外,Ru-Ni 颗粒相对尺寸大小[57]、浸渍次序也是影响催化剂活性的因素 之一。
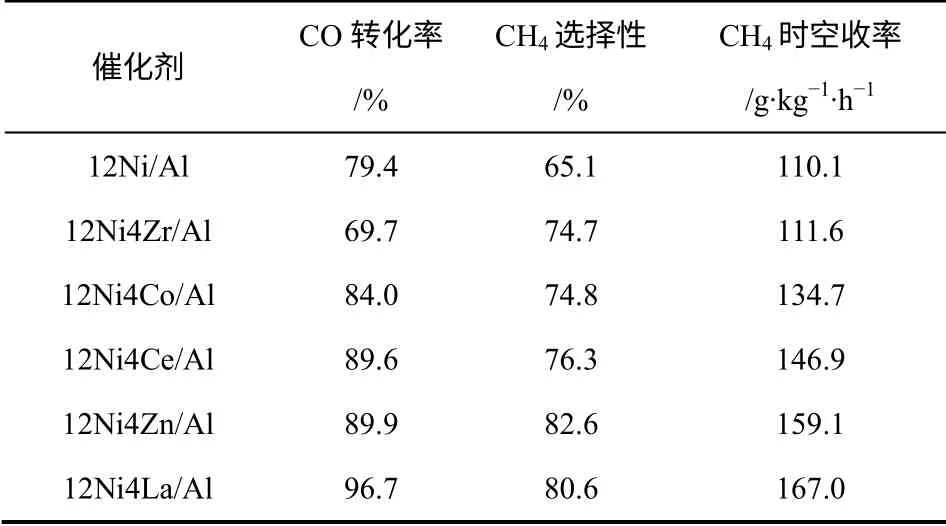
表2 不同助剂Ni 基催化剂浆态床甲烷化性能的影响[28]
此外,碱金属作为镍基催化剂助剂也有报道。王东旭等[58]总结了助剂K 对镍基催化剂积炭性能及反应活性的影响,发现助剂K 能够提高催化剂的稳定性。添加K[59]后,Ni/Al2O3催化剂的活性得到提高。添加适量Na 也可以提高Ni 的分散度,但过量的Na 反而降低了催化剂的反应活性[43]。
4 结 语
助剂改性的合成气甲烷化用镍基催化剂具有反应活性高、使用寿命长以及甲烷选择性高等优点。其改性作用机制主要体现在:①过渡金属有未充满电子的d 轨道,有利于产生电子效应,提高活性镍物种的还原度;②稀土金属助剂不但细化了活性组分镍的晶粒尺寸,提高了活性物种的分散度,而且调变了金属镍的电子状态,改善镍原子的缺电子状态;③碱土金属容易与催化剂载体形成尖晶石稳定结构,抑制了镍基催化剂的积炭和烧结,调变了催化剂的表面酸碱性,增加了对CO 的吸附能力,提高了表面活性中心数。甲烷化反应由于反应速度快、放热量大等特点,因此,载体、助剂的选取,催化剂制备方法,工艺条件等因素都会影响到甲烷化镍基催化剂的催化性能。为了开发满足工业运行的催化剂,研制的催化剂应具有反应活性高、使用周期长、抗毒能力强和生产成本低等特点,其中过渡金属、稀土金属氧化物改性的镍基催化剂甲烷化反应机理、抗中毒能力以及失活机理等方面还需要进一步的研究。此外,鉴于工业催化剂研制周期长、生产成本高等缺点,借助量子化学模拟计算辅助催化剂研发过程、使用非贵金属助剂以及复合助剂,也是近年来研究的热点。
[1] 赵亮,陈允捷. 国外甲烷化技术发展现状[J]. 化工进展,2012,31(s1):176-178.
[2] 胡大成,高加俭,贾春苗,等. 甲烷化催化剂及反应机理的研究进展[J]. 过程工程学报,2011,11(5):880-893.
[3] 赵华,魏士新,蔡进,等. 一种用于煤基合成气制代用天然气的催化剂及其制备方法:中国,102500387A[P]. 2012-06-20.
[4] Kopyscinski J,Schildhauer T J,Biollaz S M. Production of synthetic natural gas from coal and dry biomass—A technology review from 1950-2009[J]. Fuel,2010,89(8):1763-1783.
[5] Gao J J,Wang Y L,Ping Y,et al. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas[J]. RSC Adv.,2012,2:2358-2368.
[6] Wang Y Z,Li F M,Cheng H M,et al. A comparative study on the catalytic properties of high Ni-loading Ni/SiO2and low Ni-loading Ni-Ce/SiO2for CO methanation[J]. J. Fuel Chem. Technol.,2013,41(8):972-977.
[7] Li J,Zhou L,Zhu Q S,et al. Enhanced methanation over aerogel NiCo/Al2O3catalyst in a magnetic fluidized bed[J]. Ind. Eng. Chem. Res.,2013,52(20):6647-6654.
[8] Zhang J F,Bai Y X,Zhang Q D,et al. Low-temperature methanation of syngas in slurry phase over Zr-doped Ni/γ-Al2O3catalysts prepared using different methods[J]. Fuel,2014,132:211-218.
[9] Liu J,Yu J,Su F B,et al. Intercorrelation of structure and performance of Ni-Mg/Al2O3catalysts prepared with different methods for syngas methanation[J]. Catal. Sci. Technol.,2014,4:472-481.
[10] 江琦. 负载型镍催化剂上CO2加氢甲烷化研究[J]. 天然气化工,2000,25(4):9-11.
[11] Huang Y H,Wang J J,Liu Z M,et al. Highly efficient Ni-ZrO2catalyst doped with Yb2O3for co-methanation of CO and CO2[J]. Appl. Catal. A-Gen.,2013,466:300-306.
[12] Guo C L,Wu Y Y,Qin H Y,et al. CO methanation over ZrO2/Al2O3supported Ni catalysts:A comprehensive study[J]. Fuel Process. Technol.,2014,124:61-69.
[13] Tada S,Minori D,Otsuka F,et al. Effect of Ru and Ni ratio on selective CO methanation over Ru-Ni/TiO2[J]. Fuel,2014,129:219-224.
[14] Hu X J,Yan W J,Ding W H,et al. Bifunctional palladium composite membrane for hydrogen separation and catalytic methanation[J]. Chinese J. Catal.,2013,34:1720-1729.
[15] 杨霞,郑文涛,汪国高,等. MgO 对Ni/Al2O3催化剂CO 甲烷化性能的影响[J]. 现代化工,2014,34(1):90-94.
[16] Liu H Z,Zou X J,Wang X G,et al. Effect of CeO2addition on Ni/Al2O3catalysts for methanation of carbon dioxide with hydrogen[J]. J. Nat. Gas Chem.,2012,21:703-707.
[17] 李凤梅. Ni-Ce/SiO2催化剂的制备、表征及其CO 甲烷化性能[D]. 太原:山西大学,2009.
[18] 高晓庆,王永钊,李海涛,等. Mn 助剂对Ni/γ-Al2O3催化剂CO2甲烷化性能的影响[J]. 分子催化,2011,25(1):49-54.
[19] Wang Y Z,Wu R F,Zhao Y X. Effect of ZrO2promoter on structure and catalytic activity of the Ni/SiO2catalyst for CO methanation in hydrogen-rich gases[J]. Catal. Today,2010,158:470-474.
[20] Krämer M,Stöwe K,Duisberg M,et al. The impact of dopants on the activity and selectivity of a Ni-based methanation catalyst[J]. Appl. Catal. A-Gen.,2009,369:42-52.
[21] 牛雪平,姚亦淳,郝茂荣,等. 镍基甲烷化催化剂中的助剂作用Ⅱ、重稀土氧化物添加剂的结构效应和电子效应[J]. 分子催化,1999,13(1):21-26.
[22] 新民,郝茂荣,姚亦纯,等. 镍基甲烷化催化剂中的助剂作用I、稀土氧化物添加剂的电子效应[J]. 分子催化,1990,4(4):321-328.
[23] 蔺华林,李克健,赵利军. 煤制天然气高温甲烷化催化剂研究进展[J]. 化工进展,2011,30(8):1739-1743.
[24] 张成. CO 与CO2甲烷化反应研究进展[J]. 化工进展,2007,26(9):1269-1273.
[25] 杨伯伦,李星星,伊春海,等. 合成天然气技术进展[J].化工进展,2011,30(1):110-116.
[26] 李茂华,杨博,鹿毅,等. 煤制天然气甲烷化催化剂及机理的研究进展[J]. 工业催化,2014,22(1):10-24.
[27] 张旭,孙文晶,储伟. 等离子体技术对CO2甲烷化用Ni/SiO2催化剂的改性作用[J]. 燃料化学学报,2013,41(1):96-101.
[28] 崔晓曦,孟凡会,何忠,等. 助剂对Ni 基催化剂结构及甲烷化性能的影响[J]. 无机化学学报,2014,30(2):277-283.
[29] 王晓晶,新民,牛雪平,等. Ni-LiOx/γ-Al2O3催化剂中助剂及载体电子效应的研究[J]. 内蒙古大学学报:自然科学版,1994,25(4):414-419.
[30] 杨霞,田大勇,孙守理,等. CeO 助剂对Ni 基催化剂甲烷化性能的影响[J]. 工业催化,2014,22(2):137-143.
[31] Cai M D,Wen J,Chu W,et al. Methanation of carbon dioxide on Ni/ZrO2-Al2O3catalysts:Effects of ZrO2promoter and preparation method of novel ZrO2-Al2O3carrier[J]. J. Nat. Gas Chem.,2011,20(3):318-324.
[32] 沈岳年,詹肇琪,朱怀勇,等. 低镍甲烷化催化剂中助剂作用的研究(I)稀土氧化物(La2O3)的分散作用[J]. 内蒙古大学学报:自然科学版,1982,13(4):475-483.
[33] 李凤梅,王永钊,张卓,等. Ce 助剂对Ni/SiO2催化剂CO 甲烷化活性的促进作用[J]. 工业催化,2011,19(11):70-74.
[34] 洪若瑜. 磁性纳米粒和磁性流体制备与应用[M]. 北京:化学工业出版社,2009:1-20.
[35] Yu Y,Jin G Q,Wang Y Y,et al. Synthesis of natural gas from CO methanation over SiC supported Ni-Co bimetallic catalysts[J]. Catal. Commun.,2013,31:5-10.
[36] Tian D Y,Liu Z H,Li D D,et al. Bimetallic Ni-Fe total-methanation catalyst for the production of substitute natural gas under high pressure[J]. Fuel,2013,104:224-229.
[37] Bhatia S,Belteraminik J N,Do D D. Temperature programmed analysis and its applications in catalytic systems[J]. Catal. Today,1990,7(3):309-438.
[38] 王宁,孙自瑾,王永钊,等. Ni-Fe/γ-Al2O3金属催化剂的制备及其CO 甲烷化性能研究[J]. 燃料化学学报,2011,39(3):219- 223.
[39] Kustov A L,Frey A M,Larsen K E,et al. CO methanation over supported bimetallic Ni-Fe catalysts:From computational studies towards catalyst optimization[J]. Appl. Catal. A-Gen.,2007,320:98-104.
[40] 武瑞芳,张因,王永钊,等. ZrO2助剂对Ni/SiO2催化剂CO 甲烷化催化活性及其吸附性能的影响[J]. 燃料化学学报,2009,37(5):578-582.
[41] Wang T F,Porosoff M D,Chen J G G. Effects of oxide supports on the water-gas shift reaction over Pt-Ni bimetallic catalysts:Activity and methanation inhibition[J]. Catal. Today,2014,233:61-69.
[42] Jentys A,McHugh B J,Haller G L,et al. Temperature-programmed reduction of silica-supported platinum/nickel catalysts studied by XANES[J]. J. Phys. Chem.,1992,96(3):1324-1328.
[43] 陈豫,王文灼,胡常伟,等. La、Na、Mg、Ba 对低镍甲烷化催化剂性能的影响[J]. 天然气化工:C1 化学与化工,1990(5):5-11.
[44] 田大勇,杨霞,秦绍东,等. 载体及助剂对镍基甲烷化催化剂稳定性的影响[J]. 化工进展,2012,31(s1):229-231.
[45] Zhang J Y,Xin Z,Meng X,et al. Effect of MoO3on the heat resistant performances of nickel based MCM-41 methanation catalysts[J]. Fuel,2014,116:25-33.
[46] Liu Q,Gao J J,Zhang M J,et al. Highly active and stable Ni/γ-Al2O3catalysts selectively deposited with CeO2for CO methanation[J]. RSC Adv.,2014,4:16094-16103.
[47] 刘新华,苗茵,李晓丽,等. La2O3对Ni/Al2O3甲烷化催化剂的影响[J]. 物理化学学报,1995,11(8):746-750.
[48] 谢有畅,钱民协,唐有祺. 添加剂La2O3对甲烷化催化剂中镍的分散度和热稳定性的影响[J]. 中国科学:B,1983,(9):788-795.
[49] 郭雄,卿涛,韩续良,等. 一种焦炉气甲烷化催化剂及其制备方法:中国,101391218A[P]. 2009-03-05.
[50] Aksoylu A E,Msrl Z,Önsana Z I. Interaction between nickel and molybdenum in Ni-Mo/Al2O3catalysts:I CO2methanation and SEM-TEM studies[J]. Appl. Catal. A-Gen.,1998,168(2):385-397.
[51] 苗茵,石玉,刘新华,等. 镍基催化剂中钼的助剂作用研究[J]. 内蒙古石油化工,1997,23(2):6-7.
[52] 罗来涛,王敏炜,李凤仪,等. La2O3对Ni-Mo/γ-Al2O3催化剂CO和CO2甲烷化的影响[J]. 中国稀土学报,1999,17(2):120-124.
[53] Kimura M,Miyao T,Komori S,et al. Selective methanation of CO in hydrogen-rich gases involving large amounts of CO2over Ru-modified Ni-Al mixed oxide catalysts[J]. Appl. Catal. A-Gen.,2010,379:182-187.
[54] Schild C,Wokaun A,Baiker A. CO2Hydrogenation over Nickel/Zirconia catalysts from amorphous precursors : The mechanism of methane formation[J]. J. Phys. Chem.,1991,95:6341-6346.
[55] Bligaard T,Nørskov J K,Dahl S,et al. The Brønsted-Evans-Polanyi relation and the volcano curve in heterogeneous catalysis[J]. J. Catal.,2004,224(1):206-217.
[56] Tada S,Kikuchi R,Wada K,et al. Long-term durability of Ni/TiO2and Ru-Ni/TiO2catalysts for selective CO methanation[J]. J. Power Sources,2014,264:59-66.
[57] Takenaka S,Shimizu T,Otsuka K. Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts[J]. Int. J. Hydrogen Energ.,2004,29:1065-1073.
[58] 王东旭,肖显斌,高静,等. 助剂钾对镍基催化剂性能影响研究进展[J]. 化工进展,2014,33(3):668-672.
[59] Zielinski J,Znak L,Kaszkur Z. Evolution of metal phase in the course of CO hydrogenation on potassium promoted Ni/Al2O3catalyst[J]. Catal. Lett.,2010,136(1-2):92-95.
