氟尿嘧啶原料药的质量控制及稳定性研究
2015-07-19张伟男
严 宾,张伟男
(精华制药集团南通有限公司,江苏南通226407)
氟尿嘧啶原料药的质量控制及稳定性研究
严 宾,张伟男
(精华制药集团南通有限公司,江苏南通226407)
目的建立抗肿瘤药氟尿嘧啶原料药的质量控制方法,并考察其稳定性。方法采用高效液相色谱法测定氟尿嘧啶原料药含量和有关物质,并对其进行稳定性考察。结果建立了氟尿嘧啶原料药的质量控制方法,包括性状、鉴别、干燥失重(≤0.3%)、炽灼残渣(≤0.1%)、重金属(≤0.002%)、有关物质(≤0.5%)和含量测定(98.0%~102.0%),样品检测结果在规定的限度内,对氟尿嘧啶样品进行了影响因素试验考察,在高温、高湿条件下无明显降解,在光照5、10 d条件下含量均无明显变化,并分别在加速试验[(40±2)℃,相对湿度75%±5%]条件下进行了6个月和长期试验[(30± 2)℃,相对湿度60%±5%]条件下进行了9个月的稳定性试验,稳定性考察期内各项指标未见明显变化。结论所建立的氟尿嘧啶原料药质量控制方法重复性好、专属性强,结果准确、可靠,耐用性检测结果在规定的限度内,氟尿嘧啶原料药稳定性良好。
抗肿瘤药; 氟尿嘧啶; 质量控制; 参考标准; 色谱法,高效液相; 药物稳定性
5-氟尿嘧啶(5-Fu)是第1个根据一定设想而合成的抗代谢药物,是目前临床上应用最广泛的氟代嘧啶类抗肿瘤药物。其抗瘤谱较广,主要用于胃癌、结肠癌、直肠癌、乳腺癌、头颈部癌及绒毛膜上皮癌、恶性葡萄胎等,在肿瘤内科治疗中占有极其重要的地位[1-5]。5-Fu本身并无生物学活性,在体内先转变为一磷酸脱氧核糖氟尿嘧啶核苷及三磷酸氟尿嘧啶核苷后才能发挥作用,能在分子水平上代替正常代谢物,掺入生物大分子,干扰DNA的合成,发挥细胞毒素作用从而杀灭肿瘤细胞[6-9]。
1 材料与方法
1.1 材料
1.1.1 产品信息 产品名称:氟尿嘧啶;分子式:C4H3FN2O2;相对分子质量:130.08;化学结构式见图1。

图1 氟尿嘧啶化学结构式
1.1.2 仪器与试剂 NICOLET 380FT-IR红外分析仪、岛津LC-20AT液相色谱仪、DZF-6020真空干燥箱、氟尿嘧啶标准品(IOG371,含量:99.62%)、氟尿嘧啶原料药(自制)等。乙腈为色谱纯,其余试剂为分析纯。
1.1.3 质量标准 (1)性状:本品为白色至类白色无味结晶性粉末。(2)鉴别:本品的红外光谱图与对照品的红外光谱图一致。(3)干燥失重:取本品适量,按《中国药典》2010年版(二部)相关内容依法测定[10],干燥失重小于或等于0.5%(真空)。(4)炽灼残渣:取本品适量,置已恒重的铂坩埚中,按《中国药典》2010年版(二部)相关内容依法测定,炽灼残渣小于或等于0.1%。(5)重金属:取炽灼残渣项下遗留的残渣,按《中国药典》2010年版(二部)相关内容依法测定,含重金属小于或等于0.002%。(6)含量:按无水物计算,含氟尿嘧啶应为98.0%~102.0%。(7)有关物质:按“1.2.1”项的方法,精密量取标准溶液10 μL注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%~25%[11]。再精密量取标准溶液和供试品溶液各10 μL分别注入液相色谱仪,记录色谱至主峰保留时间的2倍,供试品溶液如显杂质峰,各杂质峰面积之和不得大于标准溶液主峰的峰面积(0.5%)。
1.2 方法
1.2.1 样品制备
1.2.1.1 标准溶液 精密称取氟尿嘧啶标准品10 mg,加超纯水溶解并稀释至100 mL,取此溶液1 mL至10 mL容量瓶中,加超纯水稀释至刻度。
1.2.1.2 样品溶液 精密称取氟尿嘧啶样品20 mg,加超纯水溶解并稀释至200 mL,取此溶液1 mL至10mL容量瓶中,加超纯水稀释至刻度。
1.2.1.3 含量测试方法 色谱柱为SymmetryRC18(5 μm,4.6 mm×250.0 mm),缓冲液为6.8 g/L的磷酸二氢钾水溶液,用5 mol/L的氢氧化钾调节pH值为5.7±0.1,流动相为乙腈∶缓冲液(5∶95),流速为1.0 mL/min,检测波长为254 nm;柱温为室温25℃;进样量为20 μL。进样顺序:第1针进空白,然后进5针标准溶液,接着进样品溶液,最后再进1针标准液,连续进样品的针数不得超过10针,多于10针则每10针之间交叉进1针标准液[相对标准偏差(RSD)≤0.73%]。系统适应性:标准溶液重复性RSD≤0.73%,拖尾因子范围0.95~1.05。按照外标法以峰面积进行计算,即得含量。
1.2.2 标准曲线测定方法 标准储备溶液:精密称取标准品10 mg,加入超纯水溶解并稀释至100 mL。分别精密吸取标准储备溶液 0.75、1.50、3.00、4.50、6.00 mL置容量瓶中,用超纯水稀释至50 mL,配制成质量浓度为1.51、3.02、6.04、9.06、12.08 μg/mL,进样量为20 μL,每个质量浓度连续进样3次。得平均峰面积。以平均峰面积值(A)对质量浓度(C)进行线性回归。
1.2.3 精密度测试方法 按1.2.1项的样品溶液配制方法配制6份相同质量浓度的标准样连续进样,计算其RSD;然后将此6份标准溶液放置2 d后由另一名化验员连续进样,计算其RSD,再将2名化验员的测试结果共12针,计算其RSD。
1.2.4 准确度测试方法 在不含氟尿嘧啶的空白溶剂(超纯水)中,加入氟尿嘧啶标准品,配置成3种氟尿嘧啶的质量浓度为7.05、10.07、13.10 μg/mL的溶液,放置至容量瓶中,进样量为20 μL。准确度测定:加入浓度为样品配置浓度,检出浓度为样品通过液相色谱检测出峰面积然后换算成浓度,回收率=检出浓度/加样浓度×100%。
1.2.5 耐用性测试方法 分别设定流动相流速为0.9﹑1.0、1.1 mL/min,计算氟尿嘧啶主峰保留时间、峰面积及拖尾因子。分别设定色谱柱温度为23﹑25﹑27℃,计算氟尿嘧啶主峰保留时间、峰面积及拖尾因子。分别采用同一生产商2个不同批号的色谱柱,计算氟尿嘧啶主峰保留时间、峰面积及拖尾因子。
1.2.6 样品测试方法 对 3批样品(批号分别为FLU1407601、FLU1407602、FLU1407603)质量控制项目(性状、鉴别、干燥失重、重金属、含量、有关物质、炽灼残渣)进行检测。
1.2.7 高温、高湿试验方法 对氟尿嘧啶样品进行高温、高湿测试,将氟尿嘧啶样品置80℃、90%的温、湿度条件下,放置3~12 d,定期于3、7、9、10、11、12 d时检测其含量变化。
1.2.8 光照试验方法 对氟尿嘧啶样品进行光照测试,将氟尿嘧啶样品开口放置于光照度为(4 500±500)Lx的光照装置内5~10 d,定期于5、10 d时检测其含量变化。
1.2.9 加速稳定性试验方法 取3批样品(批号分别为FLU1407601、FLU1407602、FLU1407603)置于加速稳定性试验条件下[(40±2)℃、相对湿度75%±5%],定期于0、1、2、3、4、6个月时分别取样并检测。
1.2.10 长期稳定性试验方法 取3批样品(批号分别为FLU1407601、FLU1407602、FLU1407603)置于长期稳定性试验条件下[(25±2)℃、相对湿度60%±5%],定期于0、3、6、9个月时分别取样并检测。
2 结 果
2.1 样品含量结果 测试 3批样品,批号分别为FLU1407601、FLU1407602、FLU1407603,含量分别为99.62%、99.60%、99.58%。
2.2 标准曲线测试结果 回归方程A=38 187C+8 501.8,回归系数R=0.999 6。结果表明1.51~12.08 μg/mL浓度范围内,呈现良好的线性关系,见图2。
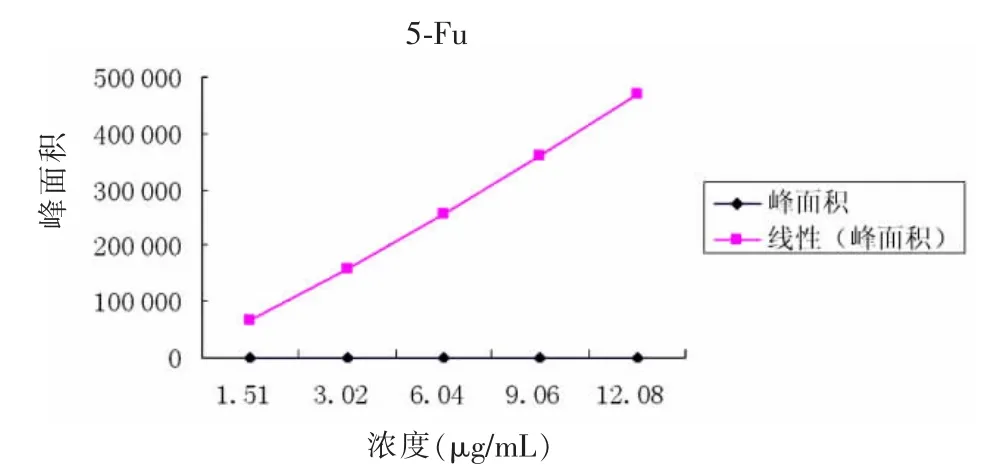
图2 峰面积-质量浓度的函数图
2.3 精密度测试结果 按照1.2.3项的方法,3次RSD结果均小于0.73,表明含量测定的进样精密度良好,满足测试要求。
2.4 准确度测试结果 准确度测试要求回收率为98.0%~102.0%,含量测定准确度结果满足测试要求,准确度良好。见表1。
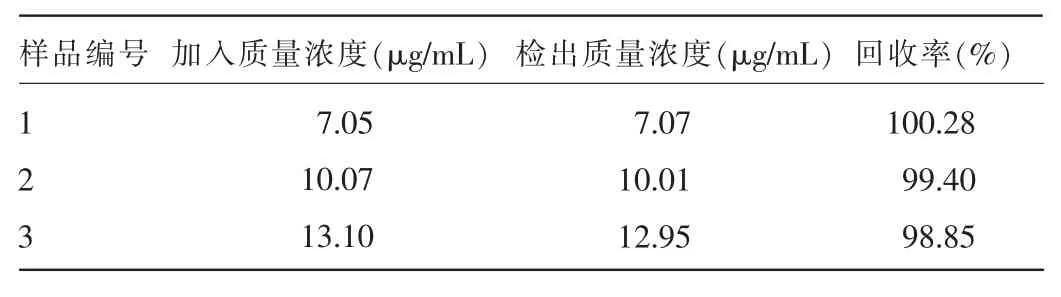
表1 准确度测试结果
2.5 耐用性测试结果 根据测试,所得结果可知流速微小变化(0.9~1.1 mL/min)、柱温微小变化(23~27℃)、同一生产商不同批号的色谱柱对含量的测定影响不大。见表2、3、4。
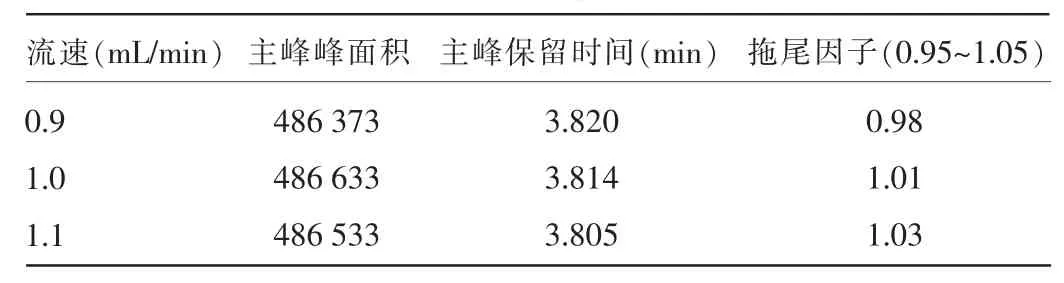
表2 流速对主峰含量影响结果

表3 色谱柱温度对主峰含量影响结果
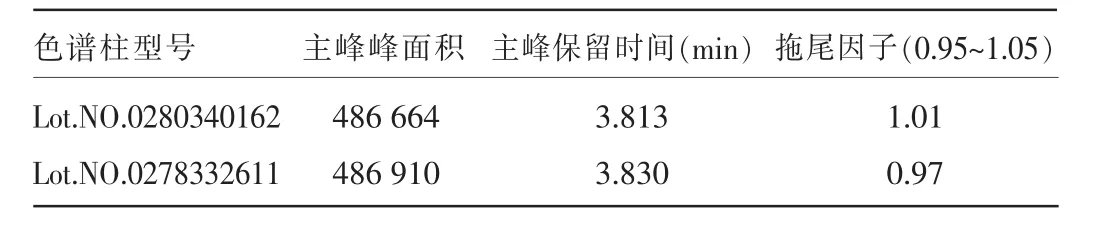
表4 色谱柱型号对主峰含量影响结果
2.6 样品测试结果 3个批次FLU1407601、FLU1407602、FLU1407603样品性状、红外光谱、干燥失重、炽灼残渣、重金属、含量、有关物质各项指标均符合质量标准。
2.7 高温、高湿试验结果 氟尿嘧啶样品在高温、高湿条件下3、7、9、10 d含量下降约1%;11、12 d含量下降约2%。表明样品在高温、高湿条件下没有明显的降解。
2.8 光照试验结果 氟尿嘧啶样品光照5、10 d含量均无明显变化。
2.9 加速稳定性试验结果 氟尿嘧啶样品在加速试验条件下1、2、3、4、6个月后质量标准中各项检测指标与0个月数据比较均无明显变化,表明本品在加速试验条件下放置6个月时质量较为稳定。
2.10 长期稳定性试验结果 氟尿嘧啶样品在长期试验条件下0、3、6、9个月后质量标准中各项检测指标与0个月数据比较均无明显变化,表明本品在长期试验条件下放置9个月时质量较为稳定。
3 讨 论
本研究根据《中国药典》(二部)对原料药的要求,探讨了氟尿嘧啶原料药的各项质量指标,建立了质量控制方法,包括性状、鉴别、干燥失重、炽灼残渣、重金属、有关物质、含量的测定,可以很好地控制氟尿嘧啶原料药产品质量,样品检测结果均在规定的限度内,表明氟尿嘧啶原料药质量可控。
本研究同时对氟尿嘧啶原料药的稳定性进行了考察,通过影响因素试验、加速稳定性试验和长期稳定性试验检测产品质量,结果提示,氟尿嘧啶样品在高温、高湿条件下没有明显的降解。在光照5、10 d条件下含量均无明显变化。加速稳定试验1、2、3、4、6个月后质量标准中各项检测指标与0个月数据比较均无明显变化,表明产品稳定性良好。长期稳定性试验3、6、9个月后质量标准中各项检测指标与0个月数据比较均无明显变化,表明产品稳定性良好。
采用高效液相色谱法测定氟尿嘧啶含量,方法简便、准确,可作为该原料药的质量控制方法,并对线性试验、精密度试验、准确度试验、耐用性试验进行方法验证,表明该控制方法重复性好,专属性强,检测结果准确、可靠,可以有效控制该产品的质量。
[1]Kugimiya N,Nishimoto A,Hosoyama T,et al.The c-MYC-ABCB5 axis plays a pivotal role in 5-fluorouracil resistance in human colon cancer cells[J].J Cell Mol Med,2015,19(7):1569-1581.
[2]Shakeel F,Haq N,Al-Dhfyan A,et al.Chemoprevention of skin cancer using low HLB surfactant nanoemulsion of 5-fluorouracil:a preliminary study[J].Drug Deliv,2015,22(4):573-580.
[3]Wu BB,Gong YP,Wu XH,et al.Fourier transform infrared spectroscopy for the distinction of MCF-7 cells treated with different concentrations of 5-fluorouracil[J].J Transl Med,2015,13:108.
[4]Matsusaka S,Lenz HJ.Pharmacogenomics of fluorouracil-based chemotherapy toxicity[J].Expert Opin Drug Metab Toxicol,2015,11(5):811-821.
[5]Kassab HJ,Khalil YI.5-Fluorouracil mucoadhesive liquid suppository formulation and evaluation[J].World J Pharm Res,2014,3(9):119-135.
[6]van Kuilenburg AB.Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil[J].Eur J Cancer,2004,40(7):939-950.
[7]Ragnhammar P,Blomgren H.How to optimize the effect of 5-fluorouracil modulated therapy in advanced colorectal cancer[J].Med Oncol,1995,12(3):187-201.
[8]Sloan KB,Wasdo S.Designing for topcial delivery:prodrugs can make the difference[J].Med Res Rev,2003,23(6):763-793.
[9]Smith NF,Figg WD,Sparreboom A.Recent advances in pharmacogenetic approaches to anticancer drug development[J].Drug Development Res,2004,62:233-253.
[10]国家药典委员会.中华人民共和国药典(2010年版:二部)[M].北京:中国医药科技出版社,2010:附录ⅥE、ⅧM、ⅧN、ⅧH。
[11]《化学药物质量标准建立的规范化过程技术指导原则》课题研究组.化学药物质量标准建立的规范化过程技术指导原则[EB/OL].[2005-6-30].http://www.doc88.com/p-3347597235110.html.
Study on quality control and stability of fluorouracil bulk drug
Yan Bin,Zhang Weinan
(Nantong Co.,Ltd.,of Jinhua Pharmaceutical Group,Nantong,Jiangsu 226407,China)
ObjectiveTo establish the quality control method of antitumor drug fluorouracil bulk drug and to investigate its stability.MethodsThe content and related substances in fluorouracil bulk drug were determined by high performance liquid chromatography(HPLC)and its stability was investigated.ResultsThe quality control method of fuorouracil bulk drug was established,including character,identification,drying loss(≤0.3%),residue on ignition(≤0.1%),heavy metals(≤0.002%),related substances(≤0.5%)and content determination(98.0%-102.0%).The sample test results were all within the limits specified.Meanwhile,the fuorouracil sample was performed the influencing factor test,the results showed that there was no obvious fluorouracil degradation under the condition of high temperature and high humidity,there were no obvious changes under the condition of 5,10 d illumination,the 6-month and 9-month stability tests were performed in the condition of the accelerated testing[(40±2)℃,relative humidity(RH)75%±5%]and the long-term testing[(30±2)℃,RH 60%±5%]respectively,the various indicators had no obvious changes during the stability investigation period.ConclusionThe established quality control method of fluorouracil bulk drug has good repeatability,strong specificity,and accurate and reliable results,the detection results of the durability test are within the limits specified,fluorouracilbulk drug has good stability.
Antineoplastic agents; Fluorouracil; Quality control; Reference standards; Chromatography,high pressure liquid; Drug stability
10.3969/j.issn.1009-5519.2015.20.009
A
1009-5519(2015)20-3070-03
2015-07-22)
严宾(1980-),男,江苏南通人,硕士研究生,工程师,主要从事医药化工研究工作;E-mail:yanbin19800918@163.com。
