Cl–…苯氰…H2O 中阴离子氢键协同效应与芳香性理论研究
2015-07-13赵光明武红娟龙泽清
宋 慧,李 斌,赵光明,武红娟,龙泽清
(中北大学化工与环境学院,太原030051)
1 引 言
共存于同一复合物的两种或多种非共价相互作用彼此增强的现象称为协同效应,它在化学反应、分子识别、生化过程的调节中发挥着极其重要的作用[1-9]. 一般来说,非共价相互作用越强,协同作用越明显. 阴离子(anion)–分子相互作用是一种较强的非共价相互作用,近年来anion…π与其它非共价相互作用间的协同效应成为人们研究的热点[10-11]. 实验表明在anion–π–π 复合物中存在anion…π 与π…π 相互作用的协同效应[12-14]. Quiñonero 等人通过理论计算发现anion…π 和氢键之间存在显著的协同效应[13]. H…X–(X=F,Cl,Br 等)阴离子氢键是一种重要的阴离子–分子相互作用[10]. 精确表征H…X–阴离子氢键的协同效应对于揭示许多生化过程至关重要[15-19]. 尽管人们已经进行了大量anion…π 协同效应的理论和实验研究,但是对于H…X–阴离子氢键协同效应鲜有报道.
众所周知,对于存在芳香环的三聚体复合物,分子间相互作用的形成会导致芳香环原子上电子的重新分配,从而使芳香性发生变化. 那么,H…X–阴离子氢键协同效应和芳香性的变化有关系吗?核独立化学位移(NICS)[20]作为一种简单可行的芳香性判据,已经被广泛应用于芳香性的表征[21]. 探索协同效应与NICS 的关系,对于揭示协同效应的本质有重要意义.
在二聚体到三聚体的变化过程中,几何结构、相互作用能、频率、偶极矩、电荷转移和化学位移的变化都已经被用来衡量协同效应的大小[22-25]. 事实上,协同效应也可以用热力学数据的变化来衡量[26]. Whitesides 等研究发现形成复合物的吉布斯自由能变ΔG 大于单体吉布斯自由能变之和时,该过程表现为正协同效应,反之为反协同效应. Gupta 和Brinkley 利用光谱方法研究正戊醇和正己醇分别与正己烷相互作用后产生的氢键协同性发现,X + Xn→Xn +1 (n≥2)氢键形成的平衡常数是2X→X2中的10 倍多[27]. 此外,已有很多关于协同效应的焓和熵的本质(ΔΔH 和-TΔΔS)[28-30]的实验研究. Subramanian等发现在DHFR…MTX…NADPH 的形成过程中存在熵驱动协同效应[31]. 大量实验和理论表明,氢键协同效应是由于熵驱动引起的[32-33].
在本文中,我们在前人对苯氰及其衍生物与水分子之间氢键相互作用的研究基础上[34-40],借助Gaussian 03 软件[41],利用量子化学从头算和密度泛函方法研究了Cl–…苯氰…H2O 三聚体模型复合物中H…X–阴离子氢键(O–H…Cl–和C–H…Cl–)与传统氢键(O – H…N 和C – H…O)之间的协同效应,探讨了协同效应与NICS 的关系,探索了其热力学协同效应的本质. 这些研究可以对芳香氰废水处理技术的改进提供理论基础.
2 计算方法
结构优化运用DFT-B3LYP 方法在6 -311 ++G(2d,p)基组水平上进行. 单点能采用B3LYP和MP2 的方法分别在6 -311 + +G(2d,p)和aug-cc-pVTZ 基组水平上计算. 借助B3LYP/6 -311 + +G(2d,p)方法进行了AIM[42]分析.
在二聚体中, 分子间相互作用能(Eint.(BN…H2O),Eint.(H2O…Cl–)或Eint.(BN…Cl–))定义为二聚体的总能量与单体(苯氰(简称“BN”),H2O 和Cl–)能量之差. 相互作用能采用基组重叠误差(BSSE)[43-44]进行了校正.
在 三 聚 体 中,E'int.(BN…H2O)、E'int.(Cl–…BN)和E'int.(Cl–…H2O)表示相近的两个单体间相互作用能.
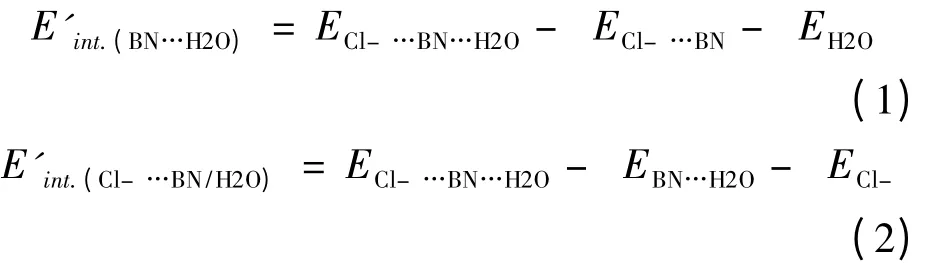
其中ECl–…BN…H2O是 指 三 聚 体 总 能 量;ECl–…BN,EBN…H2O指二聚体总能量;EH2O,ECl–指单体能量.
E″int.(H2O…BN)和E″int.(H2O…Cl–)被看成是远程相互作用能量,即复合物中较远的两个单体间相互作用能.
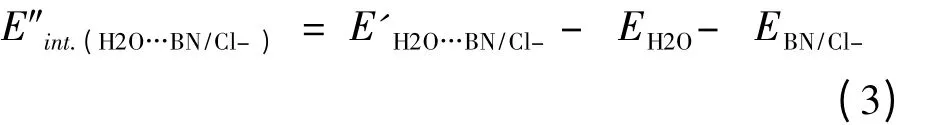
三聚体复合物的相互作用能采用下式计算:

协同效应能(Ecoop.)是指三聚体复合物的相互作用能与组成它的所有二聚体相互作用能和之差.
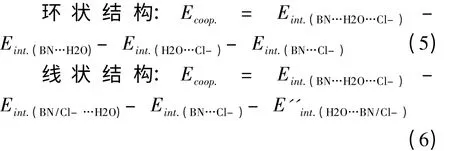

除了(3)中的E'H2O…BN/Cl–,所有公式右边能量均采用优化结构的能量.
3 结果与讨论
3.1 二聚体
在B3LYP/6 -311 + +G(2d,p)方法水平上,苯氰与H2O 形成的二聚体优化得到了六种构型,即I、II、III、IV、V 和VI,(如图1S 和2S). 在复合物I (Cs)中,苯氰分子的N 原子与水结合.在II (Cs)中,苯氰分子的N 和H 原子均与水作用. 在III (Cs),IV (Cs)和V (C2v)中,水分子的O 原子与苯氰分子的H 原子之间发生了相互作用. 在III 和V 中,O 原子分别指向苯氰的间位和对位上的H 原子;在IV 中,O 原子同时指向H原子的间位和对位. 从图2S 可看出,在B3LYP/6 -311 + +G(2d,p)水平上,H…N 和H…O 的距离在2.073 ~2.704 Å 范围内,接近于常见氢键距离,表明在I、II、III、IV、V 和VI 中形成了O–H…N 和C–H…O 氢键. 从表1S 中可看出,在B3LYP/6 -311 + +G(2d,p)水平上计算得到其相互作用能在–9.1 ~ –19.7 kJ/mol 范围内,大小顺序为II ≈I >III >IV ≈V. O–H…N 氢键相互作用比C–H…O 强.
在B3LYP/6 -311 + +G(2d,p)水平上,苯氰与Cl–形成的二聚体优化得到A 和B 两种构型(如图1S 和2S). 在A 中,Cl–同时指向苯氰的间位和对位的H 原子;在B 中,Cl–指向苯氰对位的H 原子. 在Cl–…H2O 中形成了O – H…Cl–和C–H…Cl–两种阴离子氢键.
3.2 三聚体
3.2.1 三聚体的结构
在B3LYP/6 -311 + +G(2d,p)水平上,由Cl–和I、II、III、IV 和V 构成的复合物优化后分别得到I-1 ~2、II-1、III-1 ~2、IV-1、V-1七种稳定的Cl–…苯氰…H2O 三聚体(见图1、3S和4S). 在图1 中,三聚体I - 1 (Cs)和I - 2(Cs)中同时存在O–H…N 和C–H…Cl–氢键,形成线状. 复合物II-1 (Cs),III-1 (Cs),III -2 (Cs)和V -1 (Cs)形成了环状复合物,均存在一个C–H…O 氢键和两个阴离子氢键(O–H…Cl–和C– H…Cl–). 在IV -1 中仅存在O – H…Cl–和C–H…Cl–两个阴离子氢键.
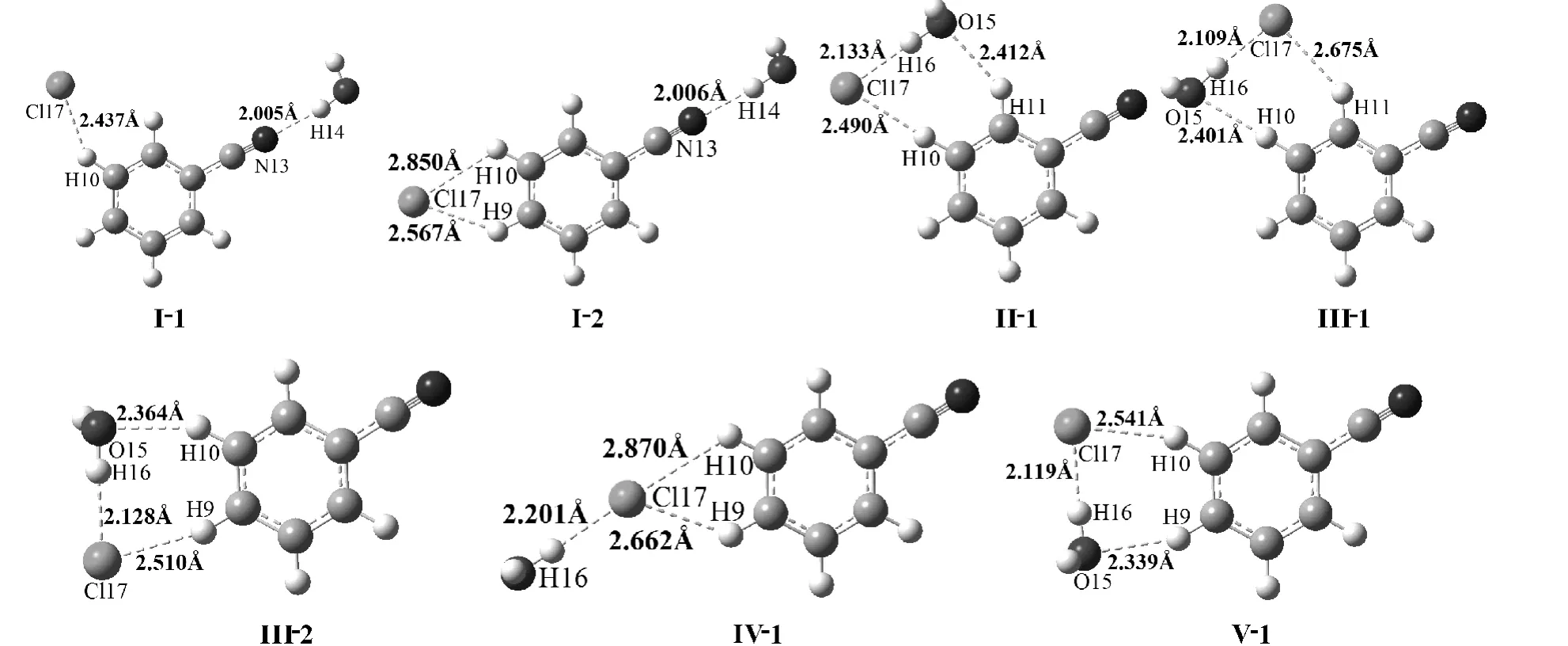
图1 B3LYP/6 -311 + +G(2d,p)方法水平上三聚体优化构形Fig. 1 The optimized geometries of ternary complexes at the B3LYP/6 -311 + +G(2d,p)level
与苯氰…H2O 二聚体比较发现,三聚体I -1和I-2 中H…N 键长以及II-1 和III-2 中H…O键长变小;III-1 中H…O 键长变大,IV-1 中的H…O 键断裂了. 这些结果表明I -1 和I -2 中O–H…N 及II-1 和III-2 中的C–H…O 的氢键相互作用增强;III -1 和IV -1 中C – H…O 氢键变弱. H…O/N 键长的显著变化表明H…Cl–阴离子氢键的形成对O– H…N 或C – H…O 氢键有较大影响,从而发生了协同效应.
此外,对于I -1、I -2、II -1 和V -1,阴离子氢键C–H…Cl–中H…Cl–的键长比相应二聚体中的键长小,表明C – H…Cl–氢键增强了.除IV-1 外,阴离子氢键O–H…Cl–中H…Cl–的键长也比相应二聚体中的键长小. 这些结果表明三聚体中O–H…Cl–氢键相互作用增强.
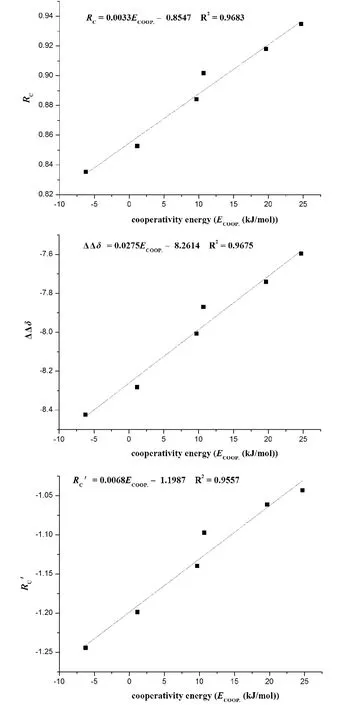
图2 协同效应能(Ecoop.)与NICS 的关系Fig. 2 The correlation between Ecoop. and NICS
从图3S 还可看出,三聚体复合物中C – CN的键长比相应二聚体中的键长小. 这表明形成三聚体后C – CN 键变强,苯环与C≡N 键的π→π*离域增强.
3.2.2 相互作用能和协同效应
表2S ~4S 和1 给出了采用B3LYP 和MP2(full)方法分别在6 -311 + +G(2d,p)和aug -cc-pVTZ 基组水平上计算得到的三聚体复合物相互作用能. 可以看出:MP2(full)方法计算得到的相互作用能比B3LYP 方法计算的结果更负;对于B3LYP 方法,从6 -311 + +G(2d,p)到aug -cc-pVTZ,相互作用能减弱;而MP2(full)方法计算的结果趋势正好相反. 因为MP2 方法在高水平基组上计算的分子间相互作用能更接近实验结果[45-47],本文除非另有说明,以下讨论均采用MP2(full)/aug-cc-pVTZ 水平上的计算结果.
从表1S ~4S 和1 中可以看出,在复合物I-1和I -2 中,E'int.(BN…H2O)和E'int.(Cl–…BN)的值比相应的苯氰…H2O 和苯氰…Cl–二聚体复合物相互作用能大. 这表明在三聚体复合物结构中,O –H…N 和C – H…Cl–氢键相互作用增强了. 因此,在I-1 和I -2 中发生了正协同效应,这与结构的分析结果吻合. 采用MP2(full)/aug -cc -pVTZ 方法,计算得到的三聚体中苯氰和水的相互作用能的增大值E'int.(BN…H2O)– Eint.(BN…H2O)与其在二聚体中Eint.(BN…H2O)的比值,即[E'int.(BN…H2O)–Eint.(BN…H2O)] /Eint.(BN…H2O),分 别 是 36.29% 和50.21 %;对于C – H…Cl–阴离子氢键相互作用能的相应结果分别是12.39% 和19.30 %. 表明在三聚体的形成过程中,O – H…N 氢键相互作用比C – H…Cl–阴离子氢键相互作用增强得更显著.
对于环状三聚体II-1、III-1、III-2 和V-1,四种方法水平计算得到的E'int.(BN…(H2O…Cl–))值均低于相同水平上相应二聚体中Eint.(BN…H2O)和Eint.(BN…Cl–)之和. 同时E'int.((BN…H2O)…Cl–)的值也低于相同水平上相应的二聚体中Eint.(BN…Cl–)和Eint.(H2O…Cl–)之和. 这些结果显示了反协同效应的发生. E'int.(BN…(H2O…Cl–))的 减 小 值 与 二 聚 体 中Eint.(BN…H2O)和 Eint.(BN…Cl–)总 和 之 比, 即[Eint.(BN…H2O)+ Eint.(BN…Cl–– E'int.(BN…(H2O…Cl–))] /(Eint.(BN…H2O)+ Eint.(BN…Cl–))分 别 是28.21%、21.70%、 9.90% 和 11.28%. 对 于E'int.((BN…H2O)…Cl–),相应值为19.46%、12.78%、5.87%和6.31%. 这些结果表明苯氰和H2O…Cl–相互作用能的相对减小值比Cl–和苯氰…H2O 大.
从表2S ~4S 和1 可看出,在四种方法水平上,三聚体复合物的结合能遵循III -2 > V -1>II-1 >IV -1 >I -2 >I -1 的顺序. 复合物的稳定性遵循III-2 >V-1 >II-1 >IV-1>I -2 >I -1 顺序. 由此可以看出,三聚体复合物更容易形成具有三个氢键相连的环状结构,线型结构不稳定.
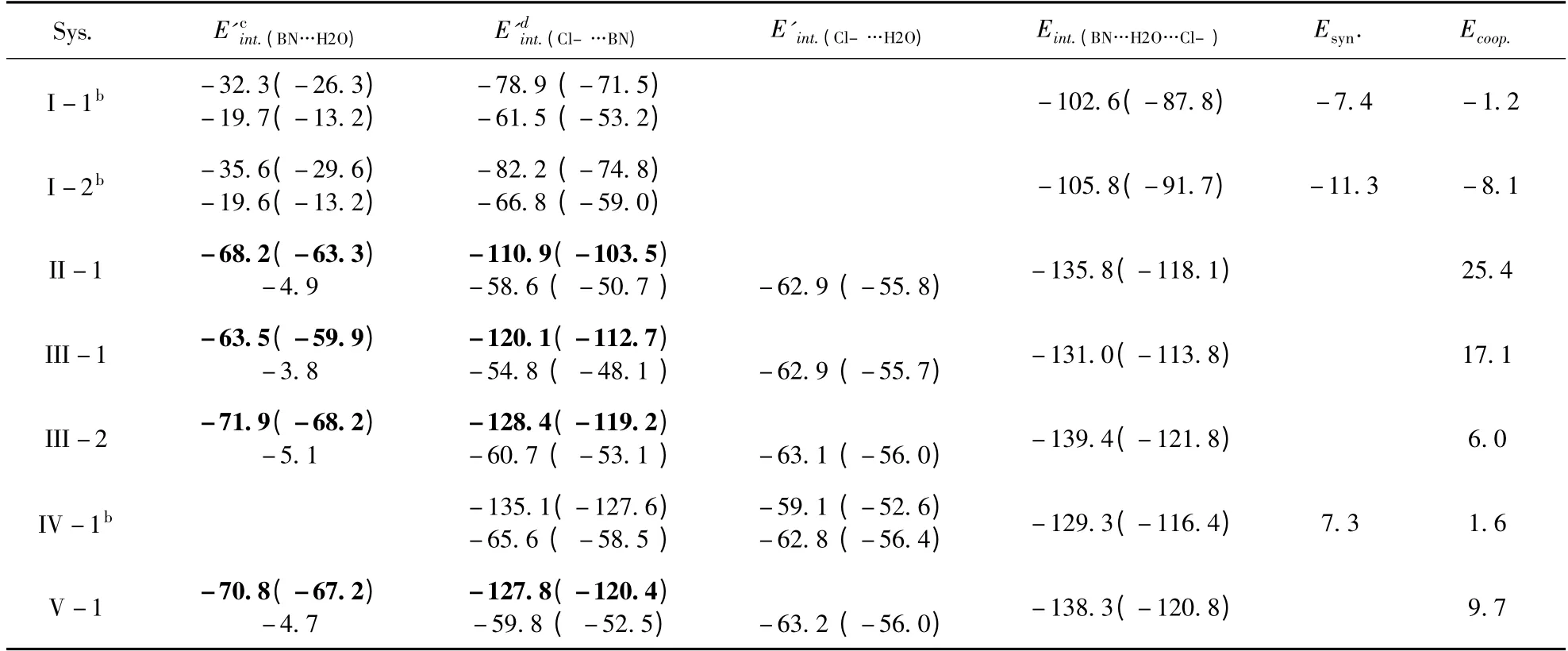
表1 MP2(full)/aug-cc-pVTZ 水平上计算三聚体分子间相互作用能(E'int.,E''int. or Eint.,kJ/mol)和协同效应能(Esyn.,Ecoop.,kJ/mol)Table 1 Interaction energies (E'int.,E''int. or Eint.,kJ/mol),synergetic energies (Esyn.,kJ/mol)and cooperativity energies(Ecoop.,kJ/mol)in the Cl–…Benzonitrile…H2O ternary system at the MP2(full)/aug-cc-pVTZ levela
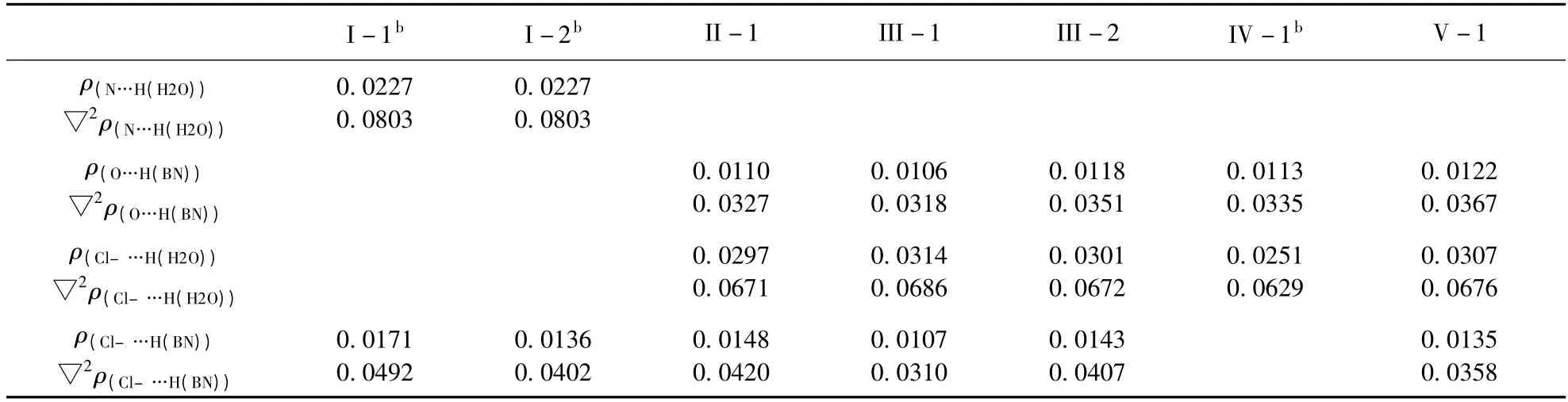
表2 B3LYP/6 -311 + +G(2d,p)水平上优化三聚体结构及AIM 结果Table 2 Selected intermolecular distances and AIM results of the ternary complexes at B3LYP/6 -311 + +G(2d,p)level
表2S ~4S 和1 给出了四种方法水平下计算得到的协同效应值. 负值表明两部分相互作用相互加强,表现为正协同效应;反之,正值则表现为反协同效应. 从表2S ~4S 和1 中可看出,对于I-1,在B3LYP/6 -311 + +G(2d,p)和B3LYP/aug-cc-pVTZ 方法水平上,其协同效应值为正;在MP2(full)/6 -311 + +G(2d,p)和MP2(full)/aug-cc-pVTZ 方法水平上是负值. 在这四种方法水平上,I-2 的协同效应值均是负值,而其他五种三聚体复合物均是正的Ecoop.值. 这些结果表明:I -2 表现出正协同效应,而II -1、III -1、III-2、IV-1 和V -1 表现出反协同效应. 这与结构和能量结果吻合. 反协同效应更容易发生在环状结构中,而正协同效应更容易发生在线性结构中. 在四种方法水平上,协同效应遵循II -1>III-1 >V-1 >III-2 >IV-1 >I-1 >I-2 的顺序.
3.3 AIM 分析
为了进一步揭示协同效应的本质,我们对二聚体和三聚体进行了AIM 分析,如图1S ~4S 和表2. 小的ρBCP值和正的▽2ρBCP值验证了O – H…N、C–H…O、O–H…Cl–和C–H…Cl–氢键相互作用的存在.
三聚体I-1 和I-2 中ρBCP(N…H)的值比二聚体中ρBCP(N…H)的值大. 这表明,三聚体中O – H…N 的 相 互 作 用 增 强. 此 外, ρBCP(Cl–…H)和ρRCP(C3,H10,Cl17,H9,C4)也均比二聚体A 大,表明三聚体中C– H…Cl–阴离子氢键增强了. 这些结果佐证了O–H…N 和C–H…Cl–的协同效应.
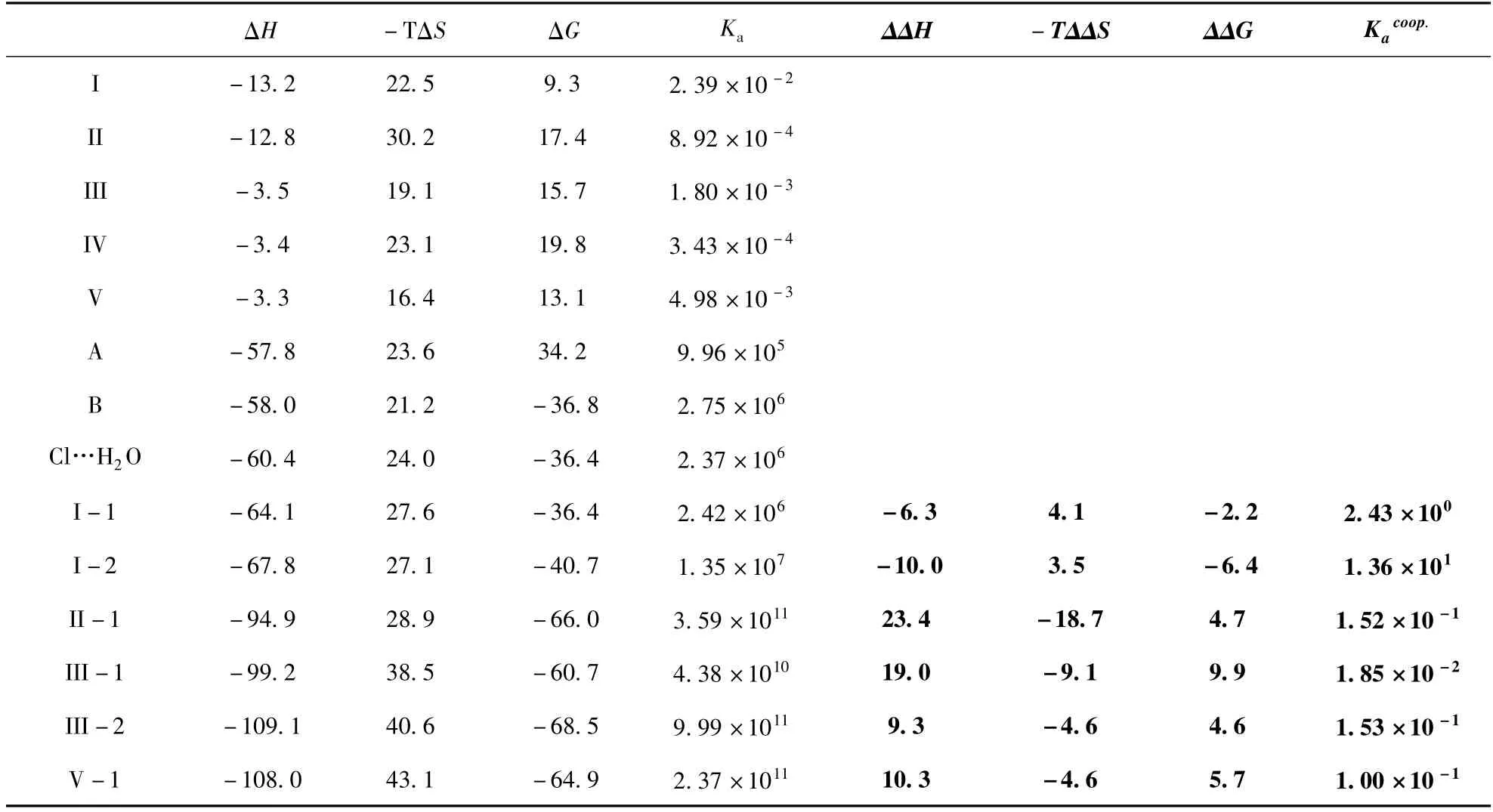
表3 B3LYP/6 -311 + +G(2d,p)水平上计算的热力学数据(kJ/mol),平衡常数(Ka)和成键常数(Kacoop.)Table 3 Thermodynamic data (kJ/mol),equilibrium constants (Ka)and binding constants (Kacoop.)at the B3LYP/6 -311 + +G(2d,p)levela
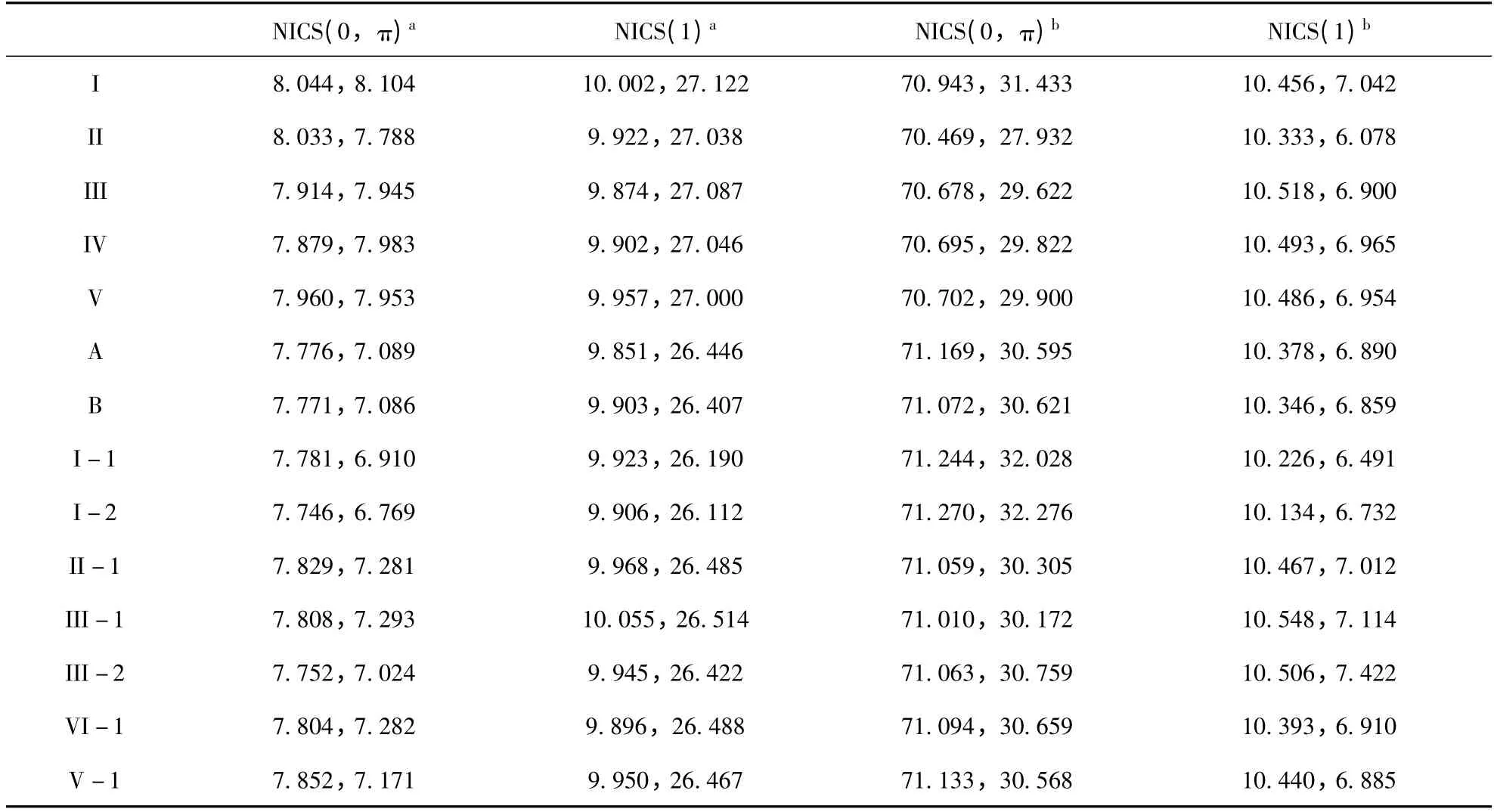
表4 B3LYP/6 -311 + +G(2d,p)水平下复合物同向性和异向性的NICS 结果(ppm)Table 4 Isotropic and anisotropic NICS (0,π)and NICS (1)(Δδ,ppm)involving the ring and C≡N bond in the binary and ternary complexes at the B3LYP/6 -311 + +G(2d,p)level
对于IV-1,与二聚体IV 和Cl–…H2O 相比,ρBCP(H…Cl–)值变小,表明O–H…Cl–和C –H…Cl–氢键变弱. 从而O– H…Cl–和C – H…Cl–之间发生了反协同效应.
对于复合物II -1、III -1、III -2、V -1,与其对应的二聚体及Cl–…H2O 比较,尽管ρBCP(O…H)和ρBCP(Cl–…H–O)的值增大了,然而连接Cl–和C–H 中H 原子的临界点消失了;而且与二聚体A 相比,其中一个C – H…Cl–阴离子氢键被破坏了. 这些结果表明,在上述四种三聚体形成过程中,虽然C – H…O 和O – H…Cl–相互作用增强了,但其中一个C – H…Cl–氢键相互作用消失了,因此发生了反协同效应.
3.4 热力学协同效应
James 等人认为,热力学协同效应可以根据吉布斯自由能变(ΔGcoop.)进行表征,ΔGcoop可以根据ΔGcoop.=ΔGter-ΔGbin计算,其中ΔGter和ΔGbin分别是指形成三聚体和二聚体过程中吉布斯自由能的变化. ΔGcoop.<0 表示热力学正协同效应,三聚体比二聚体更容易形成,三聚体中供体与接受体比二聚体中结合得更紧密[28]. 例如:Birdsall 等人发现,正协同效应可以通过控制酶抑制作用使键的结合能力大大增强,在三聚体中大约是二聚体的135 倍(相当于1 个ΔGcoop.= –2.9 kcal/mol= –RT ln135)[48]. Williams 等人也发现,热力学正协同效应会促使供体与蛋白质结合得更紧密;然而热力学反协同效应使其结构更松散[49].
在形成三聚体过程中,焓或熵贡献(ΔΔH or–TΔΔS)可以借助形成的三聚体与其对应的二聚体中ΔH 或“– TΔS”的差值来表示. ΔΔH <0和–TΔΔS<0 分别表示有利的熵和焓贡献. 研究发现,在形成生物大分子体系的复合体中,ΔΔH和– TΔΔS 是负值,而且– TΔΔS 的绝对值比ΔΔH 大,热力学协同效应主要来源于有利的熵贡献.
为了进一步探究Cl–与苯氰…H2O 形成三聚体过程的热力学协同效应的本质,采用DFT -B3LYP 方法,在6 -311 + + G(2d,p)水平上,利用统计热力学方法在298.15 K 温度下对于从分子单体到复合物过程中的ΔH,– TΔS,ΔG,ΔΔH,–TΔΔS 和ΔGcoop进行了计算. 计算方法如下:


如表3 中所示.
从表3 可以看出,苯氰…H2O(I,II,III,IV和V)二聚体复合物中,尽管焓变值(ΔH)均是负的,焓变对二聚体的形成是有利的,但– TΔS 的值是正的,且绝对值较大,导致ΔG 变为正值.因此为了减小不利的熵贡献,应该降低温度.Briand 等通过实验也发现了类似的结果[50-51]. 然而,在苯氰…Cl–二聚体(A 和B)和Cl–…H2O 形成过程中,焓变不仅是负值,而且绝对值远大于–TΔS,导致ΔG 变为负值.
在由苯氰…H2O 二聚体和Cl–形成三聚体的过程中,焓变值大大增加,其值在–64.1 ~ –109.1 kJ/mol 范围内,大约是对应的苯氰…H2O(–3.3 ~ – 13.2 kJ/mol)二聚体的6 ~10 倍.然而,熵的改变较小:在苯氰…H2O 二聚体中,不利的熵贡献(– TΔS)为16.4 ~30.2 kJ/mol,三聚体中为27.6 ~43.1 kJ/mol(在II -1 中略有减小). 因此,由于有利的焓贡献显著增加,同时不利的熵贡献增加很小,这导致在三聚体中Cl–与苯氰…H2O 的结合更牢固.
从表3 看出,对于线性结构I -1 和I -2,ΔΔH 是负值,–TΔΔS 是正值,这表明在三聚体形成过程中,熵的改变对热力学协同效应不利,而焓的改变大大促进了热力学协同效应. 换句话说,在形成三聚体复合物I -1 和I -2 中,焓变是促进热力学协同效应的重要因素. 这可能是由于三聚体复合物中有一个很强的C – H…Cl–阴离子氢键相互作用. Calderone 等人也发现在很多非共价复合物中焓贡献能促使热力学协同效应[52]. 在复合物I -1 和I -2 中,ΔGcoop值为负,表明水存在时,Cl–很容易与苯氰发生作用,且结合更加牢固,成键常数分别是二聚体的2.43 和1.36 倍.
然而,对于环状结构II-1,III-1,III-2,V-1,ΔΔH 的值为正;– TΔΔS 的值为负,这表明,在形成三聚体复合物过程中,焓变是不利的,熵变是有利的. 这表明对于这四种复合物的形成,熵变是促进热力学协同效应的一个重要的因素.Subramanian 和Kaufman 等通过实验,从热力学数据上给出了在三聚体复合物DHFR. MTX. NADPH中熵促进热力学协同效应的证据[31]. 由于│ΔΔH│比│-TΔΔS│大,ΔGcoop的值就变成了正值,导致了反协同效应,这与其结构能量的分析是一致的. 因此,三聚体复合物的形成是很困难的,这四种复合物在室温不能自发的产生. 例如,对于III-1,在水存在的前提下,Cl–结合到苯氰上的Ka的值降低到了1.85 ×10-2.
应该注意的是,对于非共价复合物,当在简谐近似下计算得到振动频率,在气相实验得到的熵和在真空计算的熵没有显著的关联性. 简谐近似计算对于低频率的振动是不恰当的,这个低频率振动对振动熵贡献很大. 因此,通常ΔS 负值变大,ΔG 的正值变大. 在溶液中,情况非常复杂. 因为溶质被溶剂化后大大地限制了他们的平动和转动,这样在气相中计算平动和转动的熵值较小. 因此,ΔG 和K 的计算是定性.
总之,在形成线性三聚体结构时,有利的焓贡献大大增加,而在形成环状三聚体结构时,有力的熵贡献大大增加. 因此,在形成三聚体线性复合物时,焓变是促进热力学协同效应的重要因素,而在形成三聚体环状复合物时,熵变却变成促进热力学协同效应的重要因素. 由于生物大分子系统中的又大又长的疏水侧链,热力学协同效应主要来源于有利的熵贡献[30,53].
3.5 NICS 和C–CN 键BDE 分析
量子化学计算已经被应用来探索了氢键协同效应对核独立化学位移(NICS)的影响. 据我们所知,还没有氢键协同效应对NICS 影响的理论研究.
作为一个简单有效的研究芳香性的标准,NICS 已经被广泛应用于芳香性的表征[54]. 在本文中,借助GIAO 方法,运用B3LYP/6 -311 + +G(2d,p)方法计算NICS. 因为NICS (0,π)(在环中心)和NICS (1)(环中心上方1 Å 处)的值是表征芳香性常用的参数,因此我们研究了NICS(0,π)和NICS (1). 另外,我们也研究了C≡N键的中心NICS (0,π)和其正上方NICS (1). 各向同性和各向异性的结果如表4 所示.
在表4 中,三聚体中环中心NICS (0,π)a的各向同性和各向异性的值比其相应的苯氰…H2O二聚体的值小. 对于环中心正上方NICS (1)a的值,除了II-1,III-1 和III-2 的各向异性的值,其他所有三聚体的NICS (1)a的值也都比其相应的苯氰…H2O 二聚体的值小. 这些结果表明:三聚体复合物中苯氰环的芳香性减弱了.
有趣的是,在三聚体复合物中,对于NICS(0,π)a和NICS (1)a各向异性的值,其顺序是I-2、I-1、III -2、V -1、II -1、III -1. 除II-1 之外,NICS (0,π)a或NICS (1)a的顺序与反协同效应的增加是一致的. NICS (1)a各向异性,Rc(Rc= NICS (1)ternary/NICS (1)binary)与Ecoop.之间的相关性如图2 所示. Ecoop.是采用B3LYP/6 -311 + +G(2d,p)方法计算得到. 其相关系数为0.9683,其线性方程如下所示:
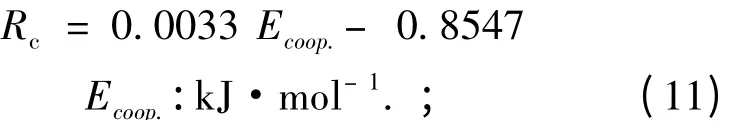
对于各向异性NICS (1)a,ΔΔδ (ΔΔδ =Δδternary– Δδbinary)与Ecoop.也得到了线性关系,如图2 所示. 其相关系数R2为0.9675. 其线性方程如下所示:
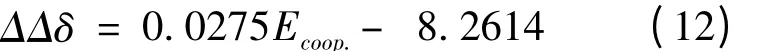
Rc' [Rc' = (NICS (1)ternary–NICS (1)binary)/NICS (1)binary]也被用于表征分子间相互作用的协同性. 可以看出Rc'与Ecoop.也有很好的线性关系,其相关系数R2可以达到0.9557,如图2 所示. 其线性方程如下:
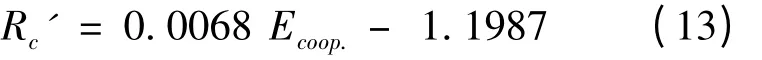
所有的结果显示:氢键的协同效应对环中心及环上方的NICS (0,π)a和NICS (1)a的值有很大的影响,这必定会导致苯氰共轭效应的变化.例如从表六可看出,三聚体C≡N 键中心各向同性和各向异性的NICS (0,π)b的值比其相应的二聚中大. 而且,三聚体中C – CN 的键长比其相应的二聚体中小.
很多的理论研究指出B3LYP 方法正确的描述了BDE 的值,而MP2 方法,由于会产生严重的自旋污染,不能精确给出BDEs[55-56]. 因此,本研究选择B3LYP/6 -311 + +G(2d,p)和B3LYP/aug-cc-pVTZ 方法计算了BDEs(见表5).
从表5 可看出,三聚体中C–CN 键的BDEs的值比其相应的二聚体中大. 这表明在三聚体复合物中C –CN 的强度增强了,这与结构的分析是一致的. 我们发现RBDE(C–CN)(BDE(C–CN)ternary/BDE(C–CN)binary)和Ecoop.之间也具有很好线性关系,如图3 所示. 其相关系数R2可以达到0.9820,他们的线性方程如下:
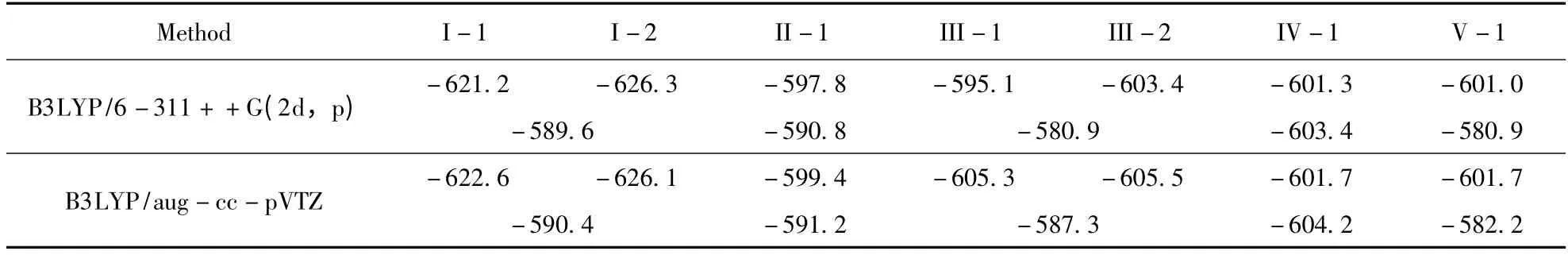
表5 苯氰…H2O 和Cl–…苯氰…H2O 中C–CN 键离解能(BDE (kJ/mol))Table 5 Bond dissociation energies (BDE (kJ/mol))of the C – CN bonds in the benzonitrile…H2O and Cl–…benzonitrile…H2O systems
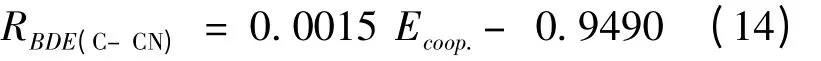

图3 协同效应能(Ecoop.)与BDE 的关系Fig. 3 The correlation between Ecoop. and BDE
4 结 论
本文采用DFT -B3LYP 和MP2(full)方法对三聚体Cl–…苯氰…H2O 中O/C – H…Cl–阴离子氢键与传统氢键O – H…N 及C – H…O 之间的协同效应、热力学性质以及芳香性进行了研究.结果表明,在线性结构中发生正协同效应,熵变是促进热力学协同效应的主要因素,而在环状结构中发生反协同效应,焓变成为主要因素. 在三聚体形成过程中,C -CN 键离解能增大、苯氰环的芳香性减弱,π→π* 共轭效应是增强的. 协同效应能 Ecoop.分别与 Rc(NICS (1)ternary/NICS(1)binary),ΔΔδ (Δδternary– Δδbinary),Rc' ((NICS(1)ternary– NICS (1)binary)/NICS (1)binary)and RBDE(C–CN)(BDE(C–CN)ternary/BDE(C–CN)binary)均具有良好的线性关系.
[1] Vijay D,Sastry G N. The cooperativity of cation -π and π - π interactions[J]. Chem. Phys. Lett.,2010,485:235.
[2] Meyer E A,Castellano R K,Diederich F. Interactions with aromatic rings in chemical and biological recognition[J]. Angew. Chem. Int. Ed.,2003,42:1210.
[3] Hesselmann A,Jansen G,Schutz M. Interaction energy contributions of H -bonded and stacked structures of the AT and GC DNA base pairs from the combined density functional theory and intermolecular perturbation theory approach[J]. J. Am. Chem. Soc.,2006,128:11730.
[4] Leist R,Frey J A,Ottiger P,et al. Nucleobase-fluorobenzene interactions:Hydrogen bonding wins over π - stacking[J]. Angew. Chem. Int. Ed.,2007,46:7449.
[5] Mignon P,Loverix S,Steyaert J,et al. Influence of the π-π interaction on the hydrogen bonding capacity of stacked DNA/RNA bases[J]. Nucl. Acids. Res.,2005,33:1779.
[6] Vijay D,Zipse H,Sastry G N. On the cooperativity of cation-π and hydrogen bonding interactions[J]. J.Phys. Chem. B,2008,112:8863.
[7] Quiñonero D,Frontera A,Escudero D,et al. MP2 study of synergistic effects between X-H/π(X = C,N,O)and π -π interactions[J]. Theor. Chem. Account.,2008,120:385.
[8] Alkorta I,Blanco F,Deyà P M,et al. Cooperativity in multiple unusual weak bonds[J]. Theor. Chem. Acta.,2010,126:1.
[9] Hunter C A,Anderson H L. What is cooperativity?[J]. Angew. Chem. Int. Ed. Engl.,2009,48:7488.
[10] Meot - Ner (Mautner)M. The ionic hydrogen bond[J]. Chem. Rev.,2005,105:213.
[11] Feng G,Qi T,Shi W,et al. A B3LYP and MP2 theoretical investigation into the synergetic effect between the O/N–H…O and O/N – H…F–anionic hydrogen-bonding interactions in N -(Hydroxymethyl)acetamide complex with F–[J]. Comput. Theor.Chem.,2013,1014:68.
[12] Lucas X,Estarellas C,Escudero D,et al. Very long-range effects:cooperativity between anion – π and hydrogen bonding interactions[J]. Chem. Phys.Chem.,2009,10:2256.
[13] Escudero D,Frontera A,Quiñonero D,et al. Interplay between anion–π and hydrogen bonding interactions[J]. J. Comput. Chem.,2009,30:75.
[14] Ebrahimi A,Masoodi H R,Khorassani M H,et al.The influence of cation – π and anion – π interactions on the strength and nature of N - H hydrogen bond[J]. Comput. Theor. Chem.,2012,988:48.
[15] Zabelin A A,Shkuropatova V A,Shuvalov V A,et al.FTIR spectroscopy of the reaction center of Chloroflexus aurantiacus:Photoreduction of the bacteriopheophytin electron acceptor[J]. Biochi. Biophys. Acta.,2011,1807:1013.
[16] Saftic' D,Žinic' B,Višnjevac A. DBU induced formation of 8 - bromoguanosine dimer with three hydrogen bonds between the GG – base pairs[J]. Tetrahedron.,2012,68:1062.
[17] Tasso B,Catto M,Nicolotti O,et al. Quinolizidinyl derivatives of bi - and tricyclic systems as potent inhibitors of acetyl - and butyrylcholinesterase with potential in Alzheimer’s disease[J]. Eur. J. Med.Chem.,2011,46:2170.
[18] Bhattacharya S,Mandal G,Ganguly T. Detailed spectroscopic investigations to reveal the nature of interaction of anionic porphyrin with calf thymus DNA[J]. J.Photoch. Photobio. B,2010,101:89.
[19] Gildenhuys S,Dobreva M,Kinsley N,et al. Arginine 15 stabilizes an SNAr reaction transition state and the binding of anionic ligands at the active site of human glutathione transferase A1 -1[J]. Biophys. Chem.,2010,146:118.
[20] von Ragué Schleyer P,Manoharan M,Wang Z X,et al. Dissected nucleus -independent chemical shift analysis of π - aromaticity and antiaromaticity[J].Org. Lett.,2001,3:2465.
[21] Hu T,Ren F,Ren J. Theoretical investigation on geometries and aromaticity of mixed boron-,nitrogenand furanoxo - containing five - membered rings B2N2OHp (p = 0 – 2)[J]. J. Mol. Struc.(THEOCHEM),2009,909:13.
[22] Kleeberg H,Klein D,Luck WAP. Quantitative infrared spectroscopic investigations of hydrogen-bond cooperativity[J]. J. Phys. Chem.,1987,91:3200.
[23] Ojamäe L,Hermansson K. Ab initio study of cooperativity in water chains:Binding energies and anharmonic frequencies[J]. J. Phys. Chem.,1994,98:4271.
[24] Wu Y D,Zhao Y L. A theoretical study on the origin of cooperativity in the formation of 310 - and α-Helices[J]. J. Am. Chem. Soc.,2001,123:5313.
[25] Kar T,Scheiner S. Comparison of cooperativity in CH…O and OH…O hydrogen bonds[J]. J. Phys.Chem. A,2004,108:9161.
[26] Cera E D. Site-specific thermodynamics:Understanding cooperativity in molecular recognition[J]. Chem.Rev.,1998,98:1563.
[27] Whitesides G M,Krishnamurthy V M. Designing ligands to bind proteins[J]. Q. Rev. Biophys.,2005,38:385.
[28] James F,Berry B,Nadezhda V K,et al. NMR structures of Apo L. casei dihydrofolate reductase and its complexes with trimethoprim and NADPH:Contributions to positive cooperative binding from ligand - induced refolding,conformational changes,and interligand hydrophobic interactions[J]. Biochemistry.,2011,50:3609.
[29] Jencks W P. On the attribution and additivity of binding energies[J]. Proc. Natl. Acad. Sci. USA,1981,78:4046.
[30] Andrea F,Jack G. Enthalpy – entropy compensation and cooperativity as thermodynamic epiphenomena of structural flexibility in ligand – receptor interactions[J]. J. Mol. Biol.,2012,417:454.
[31] Subramanian S,Kaufman B T. [J]. Proc. Natl.Acad. Sci. USA,1978,75:3201.
[32] Behrouzi R,Roh J H,Kilburn D,et al. Cooperative tertiary interaction network guides RNA folding[J].Cell.,2012,149:348.
[33] Jaroslav K,Jirˇí D. Cooperative hydrogen bonds of macromolecules. 3. A model study of the proximity effect[J]. J. Phys. Chem. B,2007,111:6118.
[34] Andrews S S,Boxer S G. Vibrational stark effects of nitriles I. Methods and experimental results[J]. J.Phys. Chem. A,2000,104:11853.
[35] Huang C Y,Wang T,Gai F. Temperature dependence of the CN stretching vibration of a nitrile -derivatized phenylalanine in water[J]. Chem. Phys. Lett.,2003,371:731.
[36] Aschaffenburg D J,Moog R S. Probing hydrogen bonding environments:Solvatochromic effects on the CN vibration of benzonitrile[J]. J. Phys. Chem. B,2009,113:12736.
[37] Kryachko E S,Nguyen M T. Hydrogen bonding in benzonitrile - water complexes[J]. J. Chem. Phys.,2001,115:833.
[38] Attah I K,Hamid A M,Meot-Ner (Mautner)M,et al. Substituent effects on noncovalent bonds:Complexes of ionized benzene derivatives with hydrogen cyanide[J]. J. Phys. Chem. A,2013,117:10588.
[39] Shan Y Y,Ren X H,Wang H J,et al. A theoretical study of the interactions between N,N - dimethylformamide and aromatic hydrocarbons[J]. Struct.Chem.,2007,18:709.
[40] Wang D,Hao C,Wang S,et al. A theoretical forecast of the hydrogen bond changes in the electronic excited state for BN and its derivatives[J]. Cent. Eur. J.Phys.,2012,10:116.
[41] Frisch M J,Trucks G W,Schlegel H B,et al. Gaussian03 Revision B. 03. Pittsburgh,PA:Gaussian,Inc.,2003.
[42] Bader R F W. Atoms in molecules,a quantum theory[M]. UK:Oxford University Press,1990.
[43] Duijineveldt F B,Duijineveldt - van de Rijdt J G C M,Lenthe J H V. State of the art in counterpoise theory[J]. Chem. Rev.,1994,94:1873.
[44] Boys S F,Bernardi F. The calculation of small molecular interactions by the difference of separate total energies. Some procedures with reduced errors[J]. Mol.Phys.,1970,19:553.
[45] Heβelmann A,Jansen G,Schütz M. Density -functional theory -symmetry -adapted intermolecular perturbation theory with density fitting:A new efficient method to study intermolecular interaction energies[J]. J. Chem. Phys.,2005,122:014103.
[46] Poovathinthodiyil R,Scott LW. Cooperative C–H…O hydrogen bonding in CO2- Lewis base complexes:Implications for solvation in supercritical CO2[J]. J.Am. Chem. Soc.,2002,124:12590.
[47] Wang Z,Zhang J,Wu J,et al. Theoretical investigation on intermolecular interactions between HCN and HNC:The nature and thermodynamic properties[J].J. Mol. Struc. (THEOCHEM),2007,806:239.
[48] Birdsall B,Burgen A S V,Roberts G C K. Binding of coenzyme analogs to Lactobacillus casei dihydrofolate reductase:binary and ternary complexes[J]. Biochemistry.,1980,19:3723.
[49] Williams D H,Stephens E,Zhou M. Ligand binding energy and catalytic efficiency from improved packing with receptors and enzymes[J]. J. Mol. Biol.,2003,329:389.
[50] Gilli R M,Sari J C,Lopez C L,et al. Comparative thermodynamic study of the interaction of some antifolates with dihydrofolate reductase. [J]. Biochim. Biophys. Acta.,1990,1040:245.
[51] Sasso S P,Gilli R M,Sari J C,et al. Thermodynamic study of dihydrofolate reductase inhibitor selectivity[J]. Biochim. Biophys. Acta.,1994,1207:74.
[52] Calderone C T,Williams D H. An enthalpic component in cooperativity:The relationship between enthalpy,entropy,and noncovalent structure in weak associations[J]. J. Am. Chem. Soc.,2001,123:6262.
[53] Nasief N N,Tan H,Kong J,et al. Water mediated ligand functional group cooperativity:The contribution of a methyl group to binding affinity is enhanced by a COO -group through changes in the structure and thermodynamics of the hydration waters of ligand-thermolysin complexes[J]. J. Med. Chem.,2012,55:8283.
[54] Wolinski K,Hilton J F,Pulay P. Efficient implementation of the gauge independent atomic orbital method for NMR chemical shift calculations[J]. J. Am.Chem. Soc.,1990,112:8251.
[55] Brink T,Haeberlin M,Jonsson M. A computational analysis of substituent effects on the O-H bond dissociation energy in phenols:polar versus radical effects[J]. J. Am. Chem. Soc.,1997,119:4239.
[56] Barckholtz C,Barckholtz T A,Hadad C M. C – H and N–H bond dissociation energies of small aromatic hydrocarbons[J]. J. Am. Chem. Soc.,1999,121:491.
