左炔诺孕酮药理活性的密度泛函研究
2015-07-10何新菊裴诗恩陈德福刘诗咏钟爱国
何新菊 裴诗恩 陈德福 刘诗咏 钟爱国

摘 要:采用密度泛函理论的DFT/B3LYP/6-311+G(d,p)方法和基组, 对左炔诺孕酮药物分子的紫外-可见光谱(UV-Vis),红外光谱(IR), 核磁共振(1HNMR)吸收光谱进行了理论模拟和指认。自然电荷(NBO)值计算表明,左炔诺孕酮分子活性最强的部位连有碳碳叁键的碳上的羟基,很可能是其发挥药理活性的亲电和亲核反应中心。
关 键 词:左炔诺孕酮;密度泛函理论;电子光谱
中图分类号:TQ 624.1 文献标识码: A 文章编号: 1671-0460(2015)09-2097-03
Abstract: Using density functional theory DFT/B3LYP/6-311 + G (d, p) method and basis set, levonorgestrels electronic spectra of UV-Vis spectroscopy, IR spectroscopy, 1HNMR spectrum and fluorescence spectroscopy were theoretically simulated and identified. Natural charge calculation shows that the hydroxyl on C atom of carbon-carbon triple bond may play the pharmacological activity center of electrophilic and nucleophilic reaction.
Key words: levonorgestrel;density functional theory;electronic spectra
口服避孕药自20 世纪50 年代诞生以来,一直被世界各国妇女认为是最有效的、可逆的、方便的避孕方式而被广泛应用[1]。鲁宾复方口服避孕药左炔诺孕酮炔雌醇片含有84个粉红色的含左炔诺孕酮0.15 mg和0.03 mg炔雌醇的活性片(active)和7个白色的惰性片(inert,无激素)[2],其中起主要作用的是左炔诺孕酮和炔雌醇。左炔诺孕酮是应用较广的一种口服避孕药。炔诺孕酮为白色或类白色的结晶性粉末;无臭,味微苦。在氯仿中溶解,在乙醇中微溶,在丙酮中略溶,在水中不溶。熔点为202~208 ℃。它可以抑制排卵并阻止孕卵着床,同时使宫颈黏液浓度增大,阻止精子前进。服用左炔诺孕酮可能会造成轻微的呕吐、恶心,以及女性下次月经周期不规则。
2005年,何淑明等探讨了左炔诺孕酮宫内缓释系统治疗子宫腺肌病的临床疗效[1,3]。左炔诺孕酮与炔雌醇可组合成复合片或三相片,有抑制排卵作用,也可用于治疗月经不调,子宫功能性出血及子宫内膜异位症等。但也容易引起月经周期不规则,头痛、胸闷[4, 5]。本文选用密度泛函方法,在优化结构的基础上, 并利用密度泛函理论,对其易发生的反应部位进行了理论预测,取得了有意义结果。
1 计算方法
用密度泛函理论(DFT)在B3LYP/6-311+G(d. p)水平上对左炔诺孕酮药物分子进行基态全优化,在优化得到的稳定结构上,采用Freq方法进行了频率分析,结果表明所有简谐振动频率全部为正值,表明其计算结果是可信的。本项目的全部计算在PC 机上完成。左炔诺孕酮(Levonorgestre, C21H28O2)的分子结构和原子编号如图1所示。通过HOMO/LUMO可以近似地判断出它的反应性,这个理论主要是基于双分子反应的分子轨道理论观察得出的三个条件:不同分子的占用轨道相互排斥;不同分子的相异电荷互相吸引;一个分子的占用轨道和另一个分子的未占轨道之间的作用导致相互吸引,尤其是HOMO和LUMO之间。据此,前线轨道理论将两种反应物的反应性简化为HOMO和LUMO的判断。
2 结果与讨论
2.1 几何结构和紫外吸收光谱
标题分子药物别名:D-甲炔诺酮、左旋18-甲炔诺酮、左旋甲基炔诺酮、左旋甲基炔诺孕酮、左旋甲炔诺酮。用gaussian 09 W程序, 在B3LYP/ STO -3G水平上进行优化计算, 得到了左炔诺孕酮的键长和部分键角值。优化的结果显示,正常的C-C单键的键长( 0.154~0.162 nm), C-O单键(0.120~0.143 nm), C=O双键(0.121~0.123 nm), O-H(0.094~1.500 nm), C=C (1.324 nm), 碳碳叁键(1.201 nm)。列出的优化后分子的部分键长数据表明, 这些键长均为正常值; C6-C13-C16-C18-C17-C14与C1-C4-C11-C13-C6-C2原子组成的六元环夹角均在109.0°附近,而C16-C19-C21-C22-C20-C18环夹角在110°~124° 附近,C1-C2-C7-C8-C3杂环夹角均在99°~106° 附近,除C3受连接的碳碳叁键影响外,其余角度没有发生明显的扭曲变形。分子中的支链与环有明显的扭转而没有处在同一平面,这样的结构受位阻效应小,将有利于药物分子发挥官能团的优势作用。
用TD DFT/B3LYP/6-311+G(d,p)方法,模拟标题分子(图2)。 分子在320 nm 处显示了紫外吸收峰,属于电子从最高占据轨道(HOMO)跃迁到最低空轨道(LUMO),这与实验UV-Vis图谱在327 nm处有最强吸收基本吻合。
2.2 NBO电荷
优化后得到了左炔诺孕酮原子的NBO电荷分布。由于O原子的电负性比C原子大,C原子的电负性比H原子大。因此, 在左炔诺孕酮分子中,2个O原子(- 0.199 e,-0.264 e)均带有净的负电荷,C原子中除C1、C3、C18、C22外均带有净的负电荷(-0.030 e ~ -0.177 e),而C1连有四个碳,C3连有3个碳和一个氧,C18连碳(有双键),C22为碳氧双键,所以C1、C3、C18、C22(0.048 e ~ 0.203 e) 均带有净的正电荷。H原子均带有正电荷。由于主环上的C原子大都带负电,致使四个碳环均带负电,具有较强的亲水性。在由电荷控制的反应中, 原子的负电荷越多, 其受亲电试剂进攻的可能性越大; 反之, 原子的正电荷越多,由于O原子的电负性比C原子大,C原子的电负性比H原子大。因此,在炔雌醇分子中,2个O原子(- 0.215 e,-0.236 e)均带有净的负电荷,C原子中除C1、C3、C20外均带有净的负电荷(-0.003 e ~ -0.223 e),而C1连有四个碳,C3连有3个碳和一个氧,C20连有C=O双键,所以C1、C3、C20(0.031 e ~ 0.093 e)均带有正电荷。H原子均带有正电荷。由于主环上的C原子大都带负电,致使四个碳环均带负电,具有较强的亲水性。由在电荷控制的反应中, 原子的负电荷越多, 其受亲电试剂进攻的可能性越大[5]; 反之, 原子的正电荷越多, 则受亲核试剂进攻的可能性也越大,因此推测C=O上的O44-OH上的O10原子和烷基上的C5原子很有可能是亲电反应的作用点。受亲核试剂进攻的可能性也越大,因此推测C=O上的O23-OH上的O10原子很有可能是亲电反应的作用点。
2.3 红外光谱和核磁共振吸收谱
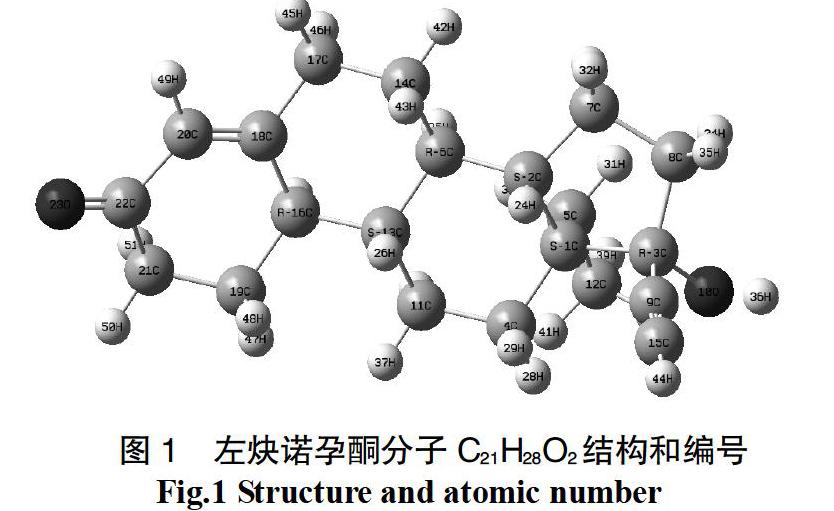
红外吸收光谱对研究分子的结构、化学键、表征和鉴别物质等有十分重要的作用。在结构优化的基础上, 在相同的计算水平上计算了左炔诺孕酮的振动红外光谱。左炔诺孕酮有51个原子,对应有147个振动模式。理论计算所得到的部分特征振动频率(0~4 000 cm-1)及其红外强度;在图3中出现了左炔诺孕酮的特征峰值750cm-1(碳碳叁键的弯曲振动),3 690 cm-1(O-H,伸缩),2 373 cm-1(碳碳叁键,伸缩)1 803 cm-1(C=C,伸缩),在低频率范围内(1 362~1 699 cm-1)还出现较多吸收峰,这主要由于碳环的H原子特征指纹区。炔雌醇分子有46个原子,对应有132个振动模式。理论计算所得到的部分特征振动频率(0~4 000 cm-1)及其红外强度列于图3中,出现了炔雌醇的特征峰值3 690 cm-1(O-H,伸缩),2 373 cm-1(碳碳叁键,伸缩),1 763 cm-1(苯环,面内伸缩), 755 cm-1(碳碳叁键的弯曲振动)在频率(730~1 381 cm-1)还出现较多吸收峰,这主要由于碳环的H原子的特征指纹区。
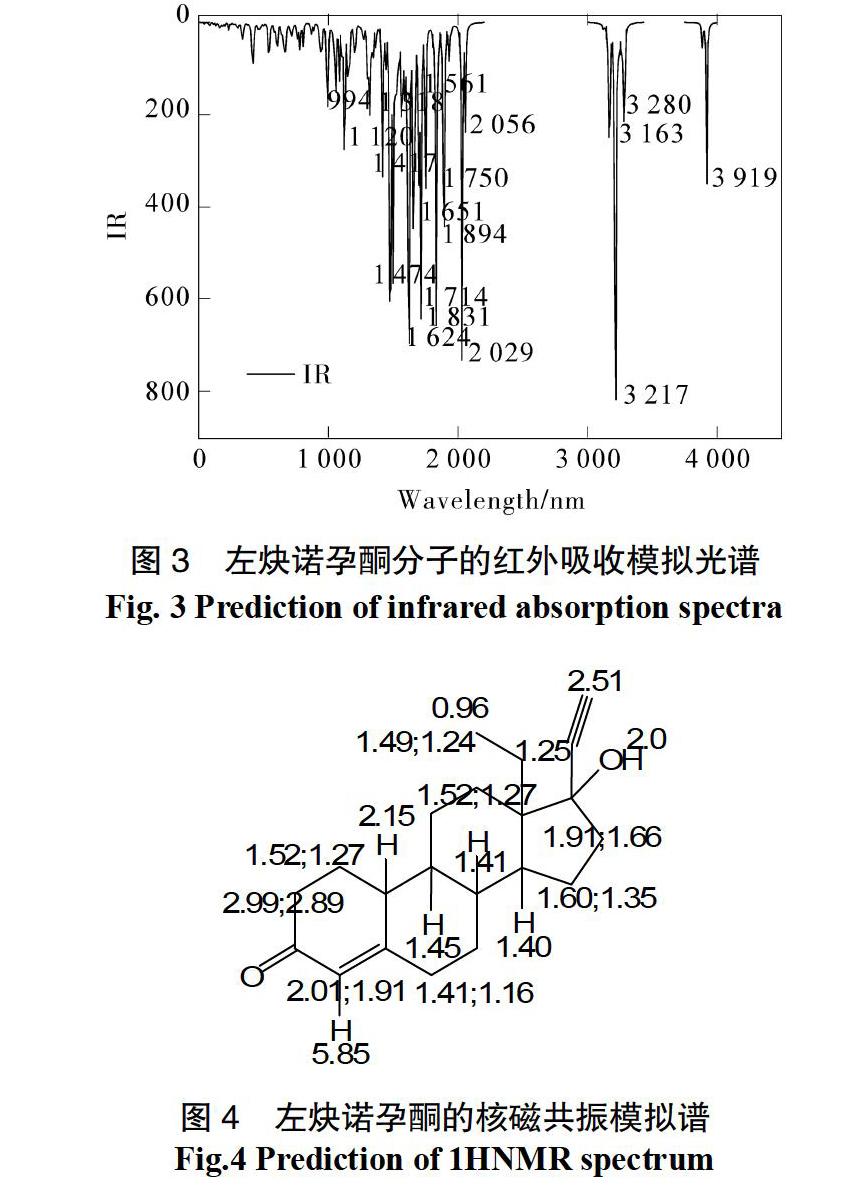
采用DFT理论模拟并指认左炔诺孕酮分子的 1HNMR(见图4)。核磁共振谱的峰特点较好地与其实验值吻合起来,也进一步验证了计算的准确性。
2.4 极性和反应活性
偶极矩和在正辛醇-水体系的分配系数(P)是用来表示药物亲脂性或疏水性的重要物理参数。左炔诺孕酮(LNG)为全合成的强效孕激素,是消旋炔诺孕酮的光学活性体,活性比炔诺孕酮强1倍,约为炔诺酮的100倍。为此,剂量比炔诺孕酮可减半,不良反应也减少。左炔诺孕酮主要作用于下丘脑和垂体,使月经中期FSH和LH水平的高峰明显降低或消失,卵巢不排卵,有明显的抗雌激素活性,比炔诺酮强10倍左右,几乎不具有雌激素活性。能使宫颈粘液变稠阻碍精子穿透。对子宫内膜转化显左炔诺孕酮示极强的孕激素活性,可使子宫内膜变薄,内膜上皮细胞呈低柱形,分泌功能不良,不利于孕卵着床。LNG也有一定雄激素活性和蛋白同化作用,口服或皮下注射均可抑制排卵。我们使用Gaussian 09W程序计算获得左炔诺孕酮和炔雌醇分子的偶极矩分别为(2.9370 D)和(0.7731 D),为弱极性。左炔诺孕酮的脂水分配系数LogP=3.06,炔雌醇的脂水分配系数logP=4, 这表明左炔诺孕酮和炔雌醇为亲脂性分子。在有机化学中,特别对芳香族化合物,确定各个原子位置在亲电或亲核取代反应的相对活性是一个重要的问题。已经提出了各种理论指标,如电荷密度分布,定域能方法等。前线轨道理论认为,最高已占分子轨道上的电子在各个原子上有一定的电荷密度分布,这个分布的大小次序决定亲电试剂进攻各个原子位置的相对难易程度,即亲电反应最易发生在HOMO最大电荷密度的原子上;与此类似,亲核反应在各个原子上发生的相对次序由LUMO的电荷密度分布决定,亲核试剂最易进攻LUMO电荷密度最大的原子。由此可知,在左炔诺孕酮中,连有碳碳叁键的碳上的羟基与C=O起药理与毒理的作用,在炔雌醇中,连有碳碳叁键的碳上的羟基与苯环上的羧基起毒理与药理的作用。
3 结 论
采用密度泛函理论的DFT/B3LYP/6-311+G(d,p)方法和基组, 对左炔诺孕酮药物分子(Levonorgestre, C21H28O2)的紫外-可见光谱(UV-Vis),红外光谱(IR), 核磁共振(1HNMR)吸收光谱进行了理论模拟和指认。
(1)几何优化表明, 左炔诺孕酮具有稳定的分子构型和良好的疏水参数,红外光谱表征与官能团吻合较准确,核磁共振与其实验值基本符合;
(2)通过NBO电荷分布以及前线轨道分析, 我们推测左炔诺孕酮分子活性最强的部位连有碳碳叁键的碳上的羟基,左炔诺孕酮将主要通过这种方式来发挥其毒理和药理作用。
参考文献:
[1]何淑明, 邢福祺. 左旋炔诺酮宫内缓释系统对子宫疾病的治疗作用[J]. 海南医学. 2006,2(4):34-36.
[2]黄 婷, 曾 佳, 曹云峰, 谢晓玉, 郑 伟, 陈良康, 陈建兴. 2007—2011年美国FDA批准上市的复方口服避孕药概况[J]. 中国新药与临床杂志, 2012,12(9):24-26.
[3]何淑明, 袁国胜, 周强, 李文. 小剂量左旋炔诺酮治疗高胆固醇血症疗效观察[J]. 郑州大学学报(医学版) , 2003, 2(6):25-26.
[4]裴泓波, 武俊青, 周 颖, 赵 瑞, 李玉艳, 张玉凤. 已婚妇女避孕方法与其人口学特征的多重对应分析[J]. 中国卫生统计, 2011,12(5) :34-36.
[5]徐海鹰, 凌峰, 徐坚, 于忠, 许轶洲, 潘浩. 左旋炔诺酮治疗80岁以上症患者的疗效[J]. 现代实用医学 , 2006,12(6):35-37.