[Fe(O)OH]⊕活化甲烷反应的理论研究
2015-07-02赵海涛
赵 慧,赵海涛
(天津大学理学院,天津 300072)
[Fe(O)OH]⊕活化甲烷反应的理论研究
赵 慧,赵海涛
(天津大学理学院,天津 300072)
采用密度泛函理论研究了气相中[Fe(O)OH]⊕与甲烷的反应机理。用B3LYP方法优化了势能面上各反应路径的过渡态和中间体等各驻点的结构,并通过振动分析和内禀反应坐标法对过渡态和中间体进行了确认,计算提出了两条可能的反应路线,揭示了[Fe(O)OH]⊕与甲烷的反应机理,同时对反应中存在的势能面交叉现象进行了研究,确定了最低势能交叉点处的作用机制。
密度泛函理论;甲烷活化;反应机理;势能面交叉现象
C-H键的活化,特别是小分子的烷烃、醇的C-H键选择性活化是现代化学中非常具有吸引力和挑战性的课题之一[1]。甲烷是煤气和天然气的主要成分。研究过渡金属催化活化甲烷以合成甲醇、甲醛等常用有机试剂,对天然气高效利用以及转化成液体原料运输具有重要的战略意义。然而,甲烷是最稳定的有机分子之一,其空间结构为正四面体,属于高度对称的Td四面体群,C-H键的离域能很高[2],约为314~523 kJ·mol-1,其4个C-H键的平均键能为414 kJ ·mol-1,要切断不活泼的C-H键是一个挑战性的课题。
近年来,由于过渡金属氧化物(TMO)在催化反应、药物开发等方面具有广泛的应用[3-4],化学家们在实验和理论上都致力于研究过渡金属氧化物参与的催化反应,希望改善或设计出更为高效的催化剂。同时,气相的实验研究能够保证一个不受外界环境干扰的实验条件,从而为化学家从严格的分子水平上揭示活化机理提供了一个理想的平台[5-7]。光谱研究表明,气相中过渡金属氧化物催化剂对烷烃中的C-H键的选择性活化具有显著的效果。1990年,Schwarz课题组首次利用傅立叶变换离子回旋共振质谱仪使用[FeO]⊕成功地将甲烷转化成甲醇[8]。同时第一行过渡金属离子[M-OH]⊕也被发现能够活化一些惰性键,如C -H键[9-10]。1986年,Cassady等[11]研究了[FeOH]⊕与甲烷的反应,证实了[FeOH]⊕可以活化甲烷C-H键。由此看来,M=O和M-OH键都可以活化C-H键。1990年,Schwarz课题组利用Fe(Ⅳ)的氢氧化物[Fe(O)OH]⊕成功活化了小分子烷烃,包括甲烷(方程a)、乙烷和丁烷[12]。

由于[FeO]⊕和[FeOH]⊕能够很好地活化甲烷C-H键,Schwarz课题组在实验上实现了[Fe(O) OH]⊕对甲烷的活化,但是从这些实验结果发现了一个问题:既然Fe=O键和Fe-OH键都能够活化甲烷C-H键,那么对于[Fe(O)OH]⊕,Fe=O键和Fe-OH键在整个活化过程中是相互促进还是某一方占主导作用呢?
为了明确Fe=O键和Fe-OH键对C-H键的活化所起的作用,作者以Schwarz课题组的实验结果为基础,采用密度泛函理论(DFT)对[Fe(O)OH]⊕和甲烷反应的机理进行了理论研究,并且对反应中可能存在的势能面交叉现象进行了讨论。以期为进一步研究甲烷活化反应提供帮助,并有助于开发具有较高活性和选择性的催化剂。
1 计算模型和理论方法
采用密度泛函理论,用B3LYP[13-15]方法tzvp[16]基组对反应过程中各势能面的反应物、过渡态、中间体和产物的几何构型进行了全参数优化。并用内禀坐标(IRC)[17-19]确认了过渡态,同时计算了反应过程中的能垒。运用Yoshizawa等[20]提出的内禀坐标垂直计算方法确定了势能面交叉点(CP点),并且运用Harvey等[21]的方法确定了最低势能交叉点(MECP点)。所有的计算都采用Gaussian 09[16]软件进行。
2 结果与讨论
2.1 反应物优化
反应物[Fe(O)OH]⊕存在5种不同自旋多重度的基态:二重态、三重态、五重态、七重态以及九重态。利用B3LYP方法在tzvp水平上优化得出这5种不同自旋多重度的反应物的结构,如图1所示。

图1 [Fe(O)OH]⊕在二重态、三重态、五重态、七重态和九重态势能面上的几何结构Fig.1 Structures of[Fe(O)OH]⊕at doublet,triplet,quintuplet,septet and nonet states
由图1可知,五重态结构能量最低最稳定,三重态和七重态次之,二重态和九重态能量分别比五重态高119.5 kJ·mol-1和341.9 kJ·mol-1。
首先讨论反应物[Fe(O)OH]⊕存在的3种较为稳定的自旋多重度的基态:三重态、五重态和七重态。其中三重态中Fe=O键长与Fe-O键长几乎相等,分别为1.662°A和1.681°A,O=Fe-O键角为149.0°,Fe-O-H键角为150.6°,能量较高;五重态结构中,Fe =O键长与Fe-O键长同样几乎相等,分别为1.730 °A和1.719°A,键长比三重态结构略长,O=Fe-O键角为149.8°,而Fe-O-H键角为162.1°,键角比三重态结构略大。五重态结构能量较三重态低3.3 kJ· mol-1。七重态相比于五重态和三重态结构,键长变长,键角增大,体系能量稍高,较五重态能量高20.1 kJ ·mol-1。在三重态、五重态和七重态结构中,Fe=O键长与Fe-O键长几乎相等,表示O=Fe-O之间存在着三中心三电子的共振离域,有效地降低了体系的能量。由于二重态和九重态结构的能量明显高于五重态结构,所以对这2个势能面上的反应不予考虑。
2.2 总观势能面
根据Schwarz课题组实验结果,[Fe(O)OH]⊕活化甲烷分别得到了P1([FeOH]⊕和CH3OH,65%)、P2 ([FeOCH3]⊕和H2O,25%)以及P3([HFeOH2]⊕和CH2O,10%)三种产物。根据实验结果,我们提出了两种可能的反应路径:path1和path2,反应在七重态、五重态和三重态势能面上各驻点的几何结构如图2所示,计算能量图如图3所示。

图2 [Fe(O)OH]⊕与CH4反应的七重态、五重态、三重态下各驻点的几何结构Fig.2 Structures of the intermediates and transition states(TSs)for the reaction of[Fe(O)OH]⊕and CH4along the reaction pathways in sep tet,quintup let and trip let states

图3 [Fe(O)OH]⊕与CH4反应的七重态、五重态、三重态势能面上的计算能量图Fig.3 Energy profiles calculated for the reaction of[Fe(O)OH]⊕and CH4along the reaction pathways in sep tet,quintup let and trip let states
2.2.1 五重态势能面上的反应
[Fe(O)OH]⊕的基态电子态为五重态,比三重态和七重态的能量分别低3.3 kJ·mol-1和20.1 kJ· mol-1。说明[Fe(O)OH]⊕是以五重态与甲烷作用进入反应路径的。首先,五重态的[Fe(O)OH]⊕和CH4通过静电吸引相互结合形成反应复合物5IM1。接着5IM1中CH4基团的一个H原子发生氢转移,由于[Fe (O)OH]⊕存在两个相互竞争的氢转移位置,反应存在两条不同的路径:path1和path2。在path1中,H原子向Fe=O双键上的O原子迁移,使得C-H键断裂同时O-H键形成。该步骤经过Fe-C-H-O四元环化的过渡态5TS1(92.0 kJ·mol-1)形成中间体5IM2。随着O=Fe-O键角逐渐增大,CH3基团中C原子向O原子(Fe=O双键上的O原子)不断靠近而形成O-C键。该步骤经过过渡态5TS2形成中间体5IM3。随后5IM3中连接Fe的CH3OH基团离去,得到实验中观测到的产物P1。整个反应是放热反应,该过程的控速步为氢转移步骤,反应能垒为92.0 kJ·mol-1。在path2中,与path1不同的是CH4基团的一个H原子向另外一个O原子(Fe-O单键上的O原子)迁移,经历过渡态5TS3,C-H键断裂并且O-H键形成,最终形成中间体5IM4。5IM4中H2O基团离去后得到实验中观测到的[FeOCH3]⊕产物。该反应也是放热反应,反应能垒为136.7 kJ·mol-1,远高于 path1控速步反应能垒,因此,path1为较为有利的反应路径。从5IM4开始,CH3基团上一个H原子向Fe金属中心迁移,同时Fe-C键断裂,剩余CH2基团向Fe=O双键上的O原子迁移,最终O-C键形成,得到中间体5IM5。5IM5中,CH2O基团离去后得到实验中观测到的产物P3。该过程的控速步同样是第一步氢转移过程,反应能垒远高于path1。所以五重态势能面上的反应path1路径比较有利。
2.2.2 七重态势能面上的反应
七重态下的反应物能量比五重态下的反应物能量高20.1 kJ·mol-1,与五重态势能面上的反应类似,第一步为形成反应复合物7IM1的过程,path1中第二步经过过渡态7TS1克服44.7 kJ·mol-1的能垒形成化合物7IM2,相比于五重态势能面上的反应,氢转移较容易进行。第三步 CH3基团的转移形成七重态中间体7IM3,随后CH3OH离去得到七重态产物[FeOH]⊕和CH3OH。path2中,从7IM1开始,克服82.8 kJ·mol-1的能垒,形成中间体7IM4,得到产物P1,随后CH3上H原子迁移至Fe原子上,C原子迁至Fe=O双键上的O原子上,形成中间体7IM5,得到产物P3。七重态势能面上path1和path2两条路径的控速步同样是氢转移过程,path1路径反应能垒远低于path2,为有利的反应路径。
2.2.3 三重态势能面上的反应
三重态势能面的反应路径与五重态非常相似,虽然三重态下的反应物能量比五重态下的反应物能量仅高出3.3 kJ·mol-1,其初始复合物以及各驻点能量均远高于五重态各驻点能量。path1中分别经过氢转移和甲基转移得到中间体3IM3,随后Fe-O键断裂得到产物P1。path2中经过一次氢转移得到中间体3IM4,随后经过氢转移Fe-C键断裂C=O形成,得到3IM5。对于三重态势能面,path1同样为有利的反应路径。
综合三个势能面的反应路径,path1为较有利的反应路径,反应经过5IM0→5IM1→5TS1→5IM2→5TS2→5IM3路线,H原子优先转移至Fe=O双键上的O原子,主要得到P1产物,与实验结果一致。反应物[Fe (O)OH]⊕中,Fe-O单键与Fe=O双键存在竞争关系,Fe=O双键起主要作用。
2.3 势能面的交叉以及最低能量交叉点处机制分析
从图3可知,path1中反应开始沿着五重态势能面的路线进行,在第一个过渡态之前和之后出现了五重态和七重态的两个交叉点,分别记为CP1和CP2,反应通过7TS1后继续沿着五重态势能面的路线进行,得到五重态产物[FeOH]⊕和CH3OH。在path2中,同样七重态的7TS3能量最低,反应存在五重态和七重态的两个交叉点,分别记为CP3和CP4,最终得到五重态的产物[FeOCH3]⊕和 H2O以及[HFeOH2]⊕和 CH2O。path1路径和path2路径都是放热的,反应的控速步均为氢转移过程,path1路线的反应能垒为84.0 kJ·mol-1,path2路线的反应能垒为 126.2 kJ·mol-1,path1为最有利的反应路线,因此实验中主要得到[FeOH]⊕和CH3OH产物。
理论研究[22-23]已经证实,两个自旋态混合时系间窜越发生的主要机制是自旋-轨道耦合作用。在path1中,CP1和CP2两个交叉点上,总自旋度的改变值分别为1和-1,满足自旋-耦合矩阵的选律(ΔS=±1),能够在自旋轨道耦合作用下使电子发生自旋翻转,在CP(MECP)点处形成系间窜越。为了深入理解CP点处的电子排布和翻转机理,我们利用NBO轨道分析和前线轨道理论以MECP1点为例进行了分析。当反应沿着path1进行到MECP1点附近时,体系存在五重态和七重态的混合。在MECP1点处五重态和七重态的前线轨道分析见图4。
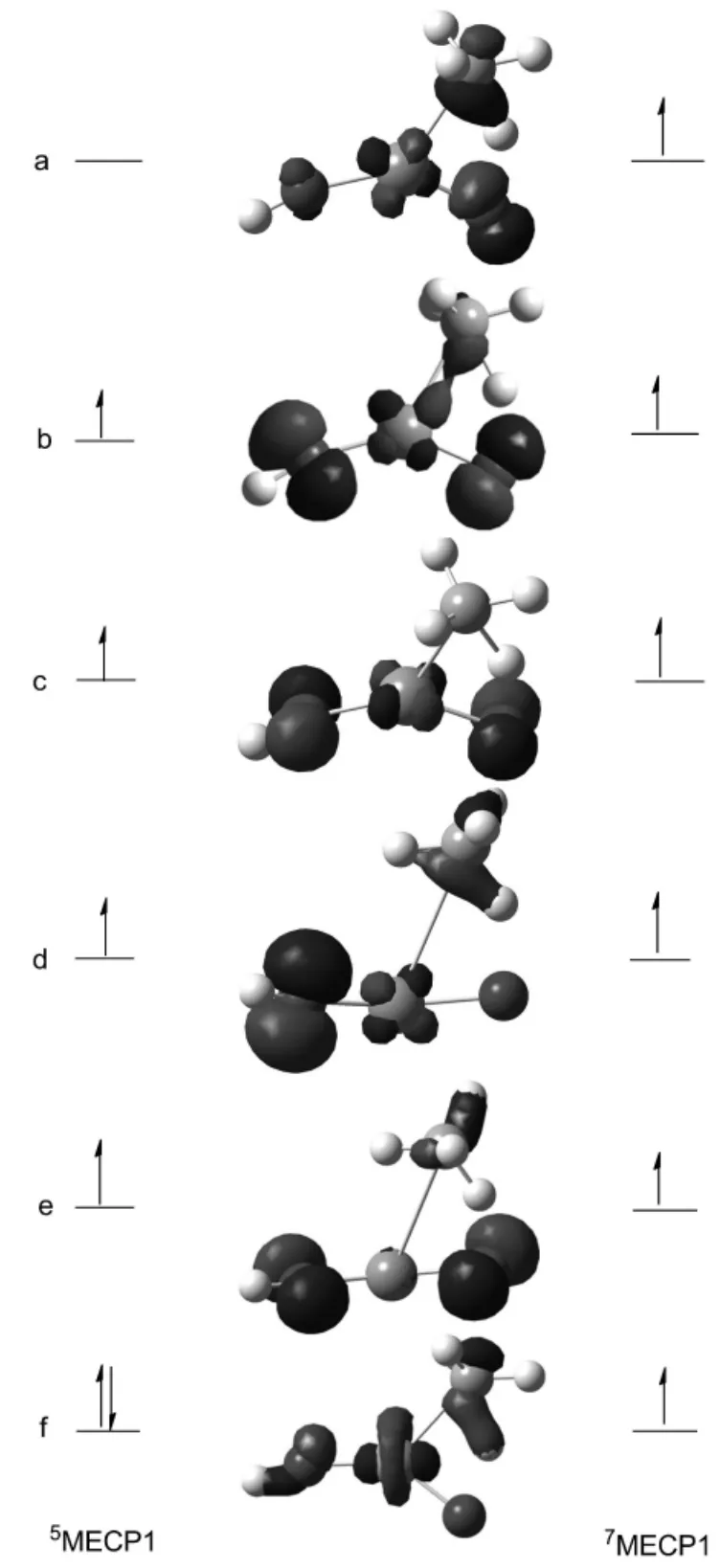
图4 MECP1点处的前线轨道分析Fig.4 The frontier orbital analysis of MECP1
由图4可知,a是dxy轨道,是七重态的最高单占轨道(HOMO),同时也是五重态的最低空轨道(LUMO)。 f为d2z轨道,是七重态的最低单占轨道(SOMO),同时也是五重态的最高双占轨道。5MECP1点处f(d2z)轨道上的一个电子会翻转到a(dxy)轨道,完成五重态向七重态的转变,由于在同一个原子内不同轨道间的电子迁移是允许的,所以该系间窜越是有效的。
3 结论
采用密度泛函理论的B3LYP/tzvp方法研究了气相中[Fe(O)OH]⊕活化甲烷生成[FeOH]⊕和CH3OH、[FeOCH3]⊕和H2O以及[HFeOH2]⊕和CH2O的反应。解释了该气相反应的反应机理,探索了3个势能面上的两条反应路径,其中生成[FeOH]⊕和CH3OH的path1路径反应能垒较低,是较为有利的反应路径,反应中Fe=O起主要作用,主要得到[FeOH]⊕和CH3OH产物,该结果与实验相符。在[Fe(O)OH]⊕活化甲烷的path1和path2两条路径中,均发生了五重态和七重态势能面交叉现象,氢转移过程中,五重态势能面向七重态势能面的跃迁降低了氢转移能垒,加快了氢转移速率。
本研究结果可以为该类反应的实验工作提供理论参考,为设计高效高选择性的催化剂,改进实验方案提供理论依据,从而推动甲烷的高效利用和甲烷化学的发展。
[1] SCHWARZH.Chemistry with methane:Concepts rather than recipes[J].Angewandte Chemie International Edition,2011,50(43): 10096-10115.
[2] GOLDEN D M,BENSON SW.Free-radical and molecule thermochemistry from studies of gas-phase iodine-atom reactions[J].Chem Rev,1969,69(1):125-134.
[3] DAY V W,KLEMPERERW G.Metal oxide chemistry in solution: The early transition metal polyoxoanions[J].Science,1985,228 (4699):533-541.
[4] SHROTRIYA V,LIG,YAO Y,et al.Transition metal oxides as the buffer layer for polymer photovoltaic cells[J].Appl Phys Lett,2006,88(7):3.
[5] BOHME D K.Gaseous ions and chemicalmass spectrometry[J].Canadian Journal of Chemistry-Revue Canadienne De Chimie 2008,86 (3):177-198.
[6] ROITHOVA J,SCHRODER D.Theorymeets experiment:Gas-phase chemistry of coinage metals[J].Coordination Chemistry Reviews,2009,253(5-6):666-677.
[7] SCHLANGEN M,SCHWARZ H.Ligand and electronic-structure effects in metal-mediated gas-phase activation of methane:A cold approach to a hot problem[J].Dalton Transactions,2009,(46): 10155-10165.
[8] SCHRODER D,SCHWARZ H.FeO⊕Activatesmethane[J].Angewandte Chemie International Edition in English,1990,29(12):1433-1434.
[9] REZABAL E,RUIPEREZ F,DE UGAL J M.Quantum chemical study of the catalytic activation ofmethane by copper oxide and copper hydroxide cations[J].Phys Chem Chem Phys,2013,15(4): 1148-1153.
[10] KRETSCHMER R,SCHLANGEN M,SCHWARZ H.Thermal activation of ammonia by transition-metal hydroxide cations[J].ChemPlus-Chem,2013,78(9):952-958.
[11] CASSADY C J,FREISER B S.Reactions of FeOH+and CoOH+with alkanes in the gas phase[J].Journal of the American Chemical Society,1986,108(19):5690-5698.
[12] SCHRODER D,SCHWARZ H.Oxidation of alkanes by[Fe(O) OH]⊕in the gas phase—the role of iron oxidation state in C-H activations[J].Angewandte Chemie International Edition in English,1991,30(8):991-993.
[13] STEPHENSP J,DEVLIN F J,CHABALOWSKIC F,etal.Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields[J].Journal of Physical Chemistry,1994,98(45):11623-11627.
[14] LEE C,YANGW,PARR RG.Metathesis polymerization of7-oxabicyclo[2.2.1]hept-5-ene derivatives[J].American Chemical Society,1988,37(12):785-789.
[15] MIEHLICH B,SAVIN A,STOLL H,et al.Results obtained with the correlation energy density functionals[J].Chemical Physics Letters,1989,157(3):200-206.
[16] FRISCH M J,TRUCKSGW,SCHLEGELH B,etal.Gaussian 09,Revision A.02[Z].Wallingford:Gaussian Inc.,2009.
[17] FUKUI K.A formulation of the reaction coordinate[J].J Phys Chem,1970,74(23):4161-4163.
[18] ISHIDA K A K,MOROKUMA K,KOMORNICKIA.The intrinsic reaction coordinate.An ab initio calculation for HNC to HCN and H-+CH4to CH4+H-[J].JChem Phys,1977,66:2153-2156.
[19] FUKUIK A.The path of chemical reactions—the IRC approach[J]. Chem Res,1981,14(12):363-368.
[20] YOSHIZAWA K,SHIOTA Y,YAMABE T.Intrinsic reaction coordinate analysis of the conversion ofmethane tomethanol by an iron-oxo species:A study of crossing seams of potential energy surfaces[J]. The Journal of Chemical Physics,1999,111(2):538-545.
[21] HARVEY JN,ASCHIM,SCHWARZH,etal.The singletand triplet states of phenyl cation.A hybrid approach for locatingminimum energy crossing pointsbetween non-interacting potential energy surfaces[J].Theor Chem Acc,1998,99(2):95-99.
[22] DANOVICH D,SHAIK S.Spin-orbit coupling in the oxidative activation of H-H by FeO+-selection rules and reactivity effects[J]. Journal of the American Chemical Society,1997,119(7):1773-1786.
[23] BECKE A D.Density-functional exchange-energy approximation with correct asymptotic behavior[J].Physical Review A,1988,38(6): 3098-3100.
Theoretical Study on the Activation of M ethane by[Fe(O)OH]⊕
ZHAO Hui,ZHAO Hai-tao
(School of Science,Tianjin University,Tianjin 300072,China)
The detailmechanisms of activation ofmethane catalyzed by[Fe(O)OH]⊕were studied with the density functional theory(DFT).The structures of the transition states and intermediates along the reaction pathways were optimized by B3LYPmethod.The transition states and intermediateswere confirmed by vibration analysis and intrinsic reaction coordinates(IRC)method.The computational results gave two possible reaction pathways and revealed the detail mechanism according to the results of the experiments.The phenomenon of cross-reaction potential energy surfaces were studied and themechanism at theminimum cross-reaction potential energy pointwas determined.
density functional theory;activation ofmethane;reaction mechanism;phenomenon of cross-reaction potential energy surfaces
O 643.36
A
1672-5425(2015)02-0016-05
10.3969/j.issn.1672-5425.2015.02.004
国家自然科学基金资助项目(20834002)
2014-11-28
赵慧(1990-),女,山东德州人,硕士研究生,研究方向:过渡金属催化反应机理,E-mail:zhaohui123@tju.edu.cn;通讯作者:赵海涛,博士,副教授,E-mail:htzhao@tju.edu.cn。
