腺苷酸活化蛋白激酶在缺血预处理诱导的神经保护中的作用
2015-06-28田园如画周中和陈会生
田园如画,周中和,陈会生
腺苷酸活化蛋白激酶在缺血预处理诱导的神经保护中的作用
田园如画,周中和,陈会生
目的 评估腺苷酸活化蛋白激酶(AMPK)及其活化型-磷酸化AMPK(pAMPK)在缺血预处理(IPC)中的作用,通过药理学方法控制pAMPK水平,评价该通路对脑梗死面积的影响。方法 对雄性大鼠行短暂(3min)大脑中动脉闭塞处理(MCAO)诱导IPC,4h或72h后再行MCAO 90min,检测IPC后AMPK及pAMPK的水平;应用AMPK药物激动剂二甲双胍或抑制剂复合物C(CC)进行处理,观察IPC与AMPK信号传导的相关性。结果 IPC预处理(72h)可使MCAO模型大鼠大脑皮质、半球及总梗死面积明显减少(P<0.05),神经功能缺损评分(NDS)明显降低(P<0.05);单纯MCAO(90min)处理后4h可明显增加pAMPK的表达,IPC预处理(4h)可使诱导的pAMPK增加明显下调(P<0.05)。应用CC腹腔注射可减少MCAO所致的大鼠脑梗死面积;IPC预处理(72h)联合CC并不能进一步减少大鼠脑梗死面积。IPC预处理(72h)行MCAO同时给予二甲双胍,二甲双胍可明显阻断IPC诱导的大脑半球、皮层及纹状体梗死面积的减少(P<0.05)。结论 AMPK及pAMPK信号在IPC介导的神经保护作用中发挥重要作用,IPC的神经保护作用可能与下调pAMPK水平有关。
脑缺血;梗死,大脑中动脉;AMP活化蛋白激酶类;二甲双胍;复合物C
腺苷酸活化蛋白激酶(adenosine 5'-monophosphateactivated protein kinase,AMPK)是一种异源三聚体蛋白,在大多数哺乳动物组织和器官,包括大脑中均有表达[1-2]。AMPK主要由α、β、γ三个亚基组成,α亚基起催化作用,β、γ亚基主要维持三聚体的稳定性以及作用底物的特异性,它们各自有不同的组织表达和生理特性[3-4]。当能量供应不足时,AMPK可以被α亚基中172位的磷酸化苏氨酸激活[5-6],被认为是细胞能源动力的重要调控因子[3,7]。AMPK在细胞水平通过抑制三磷腺苷(ATP)的消耗代谢、激活ATP的合成代谢维持能量储备,启动级联反应保证代谢适应性和细胞活力[4,8]。缺血发生后会立即诱导pAMPK增高并持续24h,所以药物性抑制AMPK具有神经保护作用[2,9-10]。
缺血预处理(ischemic preconditioning,IPC)可导致哺乳动物的适应性耐受,因此短暂的非损伤性刺激反而可降低后续损伤的严重程度[11]。IPC可降低后续损伤中ATP的消耗速率,所以IPC的有益作用被归结于代谢抑制[12]。有研究指出,IPC激活的许多信号通路由AMPK调控,提示AMPK可能是缺血性代谢适应的重要介质[13-14]。
本研究目的在于进一步探讨AMPK信号通路在IPC中的作用。由于IPC的神经保护作用需延迟数小时才能完全显现,早期一些研究认为72h是IPC发挥神经保护作用的高峰[15]。本研究选择IPC 后72h行大脑中动脉闭塞处理(middle cerebral artery occlusion,MCAO)90min,评估AMPK对IPC的作用,并通过药物干预控制pAMPK水平,评价该方法对脑梗死面积的影响。
1 材料与方法
1.1 动物及试剂 SD雄性大鼠由沈阳军区总医院动物实验科提供,体重200~250g,实验动物的使用遵循伦理相关要求。pAMPK、AMPK、AMPK激活剂二甲双胍(Metformine )购自Cell Signaling公司,热休克蛋白70(HSP70)购自Santa Cruz公司,β-actin购自Sigma公司,AMPK、pAMPK羊抗兔IgG、HSP70、β-actin羊抗鼠IgG购自Chemicon International公司,AMPK抑制剂复合物C(CC)购自Merck-Calbiochem公司。
1.2 IPC与MCAO模型制备 雄性SD大鼠右侧行MCAO处理,将线栓抽出行再灌注,诱导局部短暂脑缺血[9,16]。假手术操作:线栓未进入大脑中动脉,其余与IPC操作相同。采用激光多普勒血流仪(LDF)测定脑血流。
1.3 实验设计及分组
1.3.1 检测IPC对梗死面积的影响 实验设IPC+MCAO组和对照(Sham)+MCAO组(n=8)。IPC+MCAO组在IPC后72h行MCAO 90min,Sham+MCAO组假手术IPC后72h行MCAO 90min。各组大鼠处理后取脑,测定大脑半球总梗死面积,并在MCAO处理24h后进行神经功能缺损(nervous functional deficiency,NDS)评分。
1.3.2 检测IPC对pMARK蛋白表达的影响 实验设5个组(n=6):IPC 4h组,IPC 72h组,Sham组,IPC+MCAO 4h组和MCAO 4h组。IPC 4h组和IPC 72h组:12只大鼠行单纯IPC,其中6只4h后取脑,6只于72h取脑。IPC+MCAO 4h组:IPC后72h行MCAO(90min),4h后取脑;MCAO 4h组:MCAO(90min)后4h取脑;Sham组:12只大鼠行假手术IPC处理。各组大鼠处理后取脑组织,待测pMARK蛋白表达。
1.3.3 检测AMPK抑制剂CC对梗死面积的影响
实验设4组(n=8):CC+MCAO组,CC溶媒生理盐水(saline)+MCAO组,IPC+CC+MCAO组,IPC+saline+MCAO组。各组处理如下:CC+MCAO组:MCAO同时给予CC(10mg/kg CC溶于0.2ml/20g生理盐水,腹腔注射);saline+MCAO组:MCAO同时给予CC溶媒生理盐水(0.2ml/20g,腹腔注射);IPC+CC+MCAO组:IPC后72h行MCAO同时给予复合物C;IPC+saline+MCAO组:IPC后72h行MCAO处理同时给予溶媒生理盐水。各组大鼠处理后取脑,测皮质、纹状体以及总梗死面积。
1.3.4 检测AMPK激活剂二甲双胍(Metformine)对梗死面积的影响 实验设3组(n=6):IPC+MCAO+二甲双胍、I PC+MC AO+二甲双胍溶媒生理盐水(saline)组,Saline+MCAO组,每组6只大鼠。处理如下:IPC+MCAO+二甲双胍组:IPC 后72h行MCAO同时给予二甲双胍(100mg/kg二甲双胍溶于0.2ml/20g生理盐水中,腹腔注射)。IPC+MCAO+saline组:IPC后72h行MCAO处理同时给予溶媒saline(0.2ml/20g,腹腔注射)。Saline+MCAO组:MCAO处组同时给予saline(0.2ml/20g,腹腔注射)。各组大鼠处理后取脑,测皮质、纹状体以及总梗死面积。
1.3.5 药物处理方式 当MCAO开始时,向腹腔内注射复合物C(10mg/kg)或二甲双胍(100mg/kg)或生理盐水,溶于生理盐水后0.2ml/20g,剂量参考文献[9,17-18]。
1.4 检测指标及检测方法 研究者在双盲情况下进行指标检测及分析。
1.4.1 行为学评分 分别于MCAO缺血期间和MCAO后24h对大鼠进行神经功能缺损评分(nervous functional deficiency,NDS)。评分标准如下:0分,无缺损症状;1分,前肢无力、提尾时躯干向同侧倾倒;2分,向患侧转圈;3分,患侧不能承受体重;4分,无自发活动或滚动[9]。研究人员在双盲情况下进行评分。
1.4.2 梗死面积分析 IPC再灌注后24h(预处理后96h),将大鼠安乐死,取脑,2mm冠状切片,1.5% 2,3,5-三苯基氯化四氮唑(TTC)染色,根据TTC染色后梗死面积与其所占切片总面积的百分比进行分析描述[9]。
1.4.3 Western blotting检测 参考既往研究方法行Western blotting分析[9]。由于pAMPK表达变化具有动态特性,断头取脑后需尽快取脑、冰冻保存,匀浆后分装保存于–80℃,避免反复冻融。IPC后4h或72h取脑。依次加入pAMPK(1:500)、AMPK(1:1000),HSP70(1:2500),β-actin(1:5000)作内参,4℃孵化过夜,再加入二抗(AMPK、pAMPK羊抗兔IgG,1:10 000,HSP70、β-actin羊抗鼠IgG,1:5000),室温下孵育45min,滴加ECL显色液,采用Imagepro Plus软件采集图像并分析。
1.5 统计学处理 采用SPSS 10.0软件进行分析。定量资料包括大脑梗死面积百分比、NDS评分、pAMPK水平,数据符合正态分布采用±s描述,多组比较采用单因素方差分析,进一步两两比较采用SNK-q检验;不符合正态分布,采用25%中位数进行描述,组间比较采用秩和检验。P>0.05为差异有统计学意义。
2 结 果
2.1 MCAO模型确定 MCAO处理后,用激光多普勒血流仪(LDF)测定脑血流,证实动物脑血流量下降≥80%,再灌注时又恢复正常,提示造模成功。
2.2 IPC对脑梗死及NDS评分的影响 经3min M C AO后诱导I P C刺激的鼠脑组织在T TC染色时未见损伤。MCAO前72h行IPC可明显减少动物大脑半球总梗死面积(sham+MCAO vs IPC+MCAO, 44.9±3.2% vs 32.8±3.1%,P<0.05,图1A、B)。同时,IPC也降低了MCAO后的NDS评分(Sham+MCAO vs IPC+MCAO,3.00 vs 2.01,P<0.05;图1C)。IPC未导致梗死,可诱导神经保护作用、改善行为障碍。
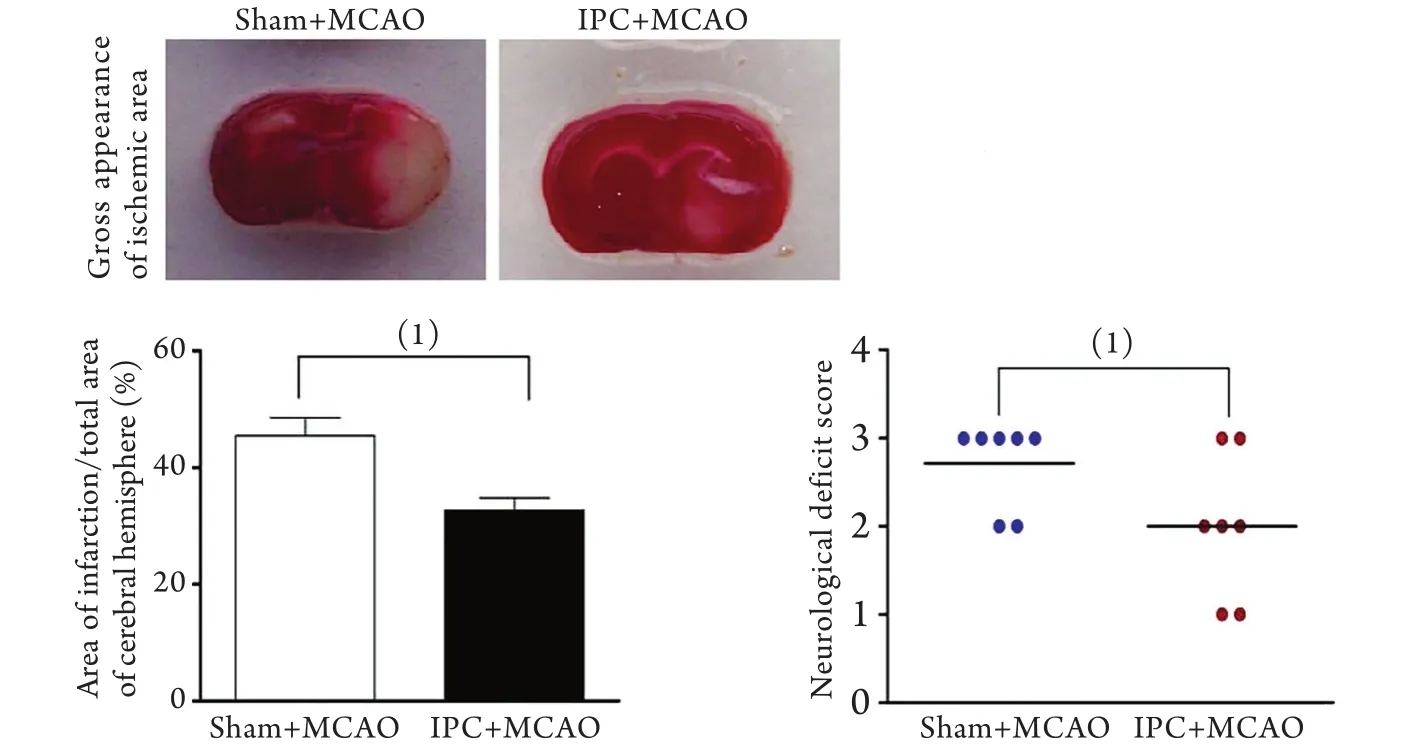
图1 Sham+MCAO与IPC+MCAO组梗死面积和NDS评分比较Fig.1 Comparison of the infarct area and NDS between sham+MCAO group and IPC+MCAO group

图2 各组AMPK和pAMPK表达及比较Fig. 2 Expression and comparison of AMPK and pAMPK levels among sham, IPC, sham+MCAO and IPC+MCAO groups
2.3 IPC对pAMPK蛋白表达的影响 由图2可见,单纯IPC后4h,pAMPK水平可明显提高(Sham vsIPC,0.9±0.2 vs 1.6±0.1,P<0.05),但在72h,IPC组大鼠的pAMPK较sham组有所降低(Sham vs IPC,1.2±0.2 vs 1.3±0.1,P<0.05)。单纯MCAO(90min)处理后4h可明显增加pAMPK的表达(MCAO 4h vs IPC 4h vs Sham,2.3±0.2 vs 1.6±0.1 vs 0.9±0.2,P<0.05)。进一步观察显示,预先IPC处理可明显下调pAMPK的表达(Sham+MCAO vs IPC+MCAO,2.3±0.2 vs 1.2±0.1;P<0.05)。由于sham组pAMPK水平无明显改变,因此将所有Sham组的pAMPK数值标准化(图2)。该结果与既往研究证实卒中可提高pAMPK表达[17]相符合。
2.4 CC治疗组缺乏预处理效应 首先探讨MCAO同时给予大鼠AMPK抑制剂CC腹腔注射对梗死面积的影响。实验中,saline+MCAO组1只大鼠死亡,7只大鼠数据进入统计。与saline组相比,CC处理可明显减少梗死面积(saline+MCAO vs CC+MCAO,总梗死面积46.8±3.6 vs 33.3±2.6,P<0.05;皮层58.7±3.6 vs 36.9±3.0,P<0.05;纹状体69.5±2.7 vs 56.0±2.8,P<0.05)。
进一步观察I P C是否与CC注射有协同作用。与IPC+saline处理比较,IPC与CC协同并未能进一步减少梗死面积(IPC+CC+MCAO vs IPC+saline+MCAO,总梗死面积31.4±1.4 vs 32.2±2.4,P>0.05;皮层35.7±3.2 vs 37.8±1.4, P>0.05;纹状体54.8±3.1 vs 56.2±2.5,P>0.05,图3)。
2.5 二甲双胍对神经保护作用的影响 大鼠行IPC后,在MCAO处理的同时又给予动物腹腔注射AMPK激活剂二甲双胍。结果发现,与saline对照组比较,二甲双胍可消除IPC诱导的神经保护作用(IPC+saline+MCAO vs IPC+Metformin+MCAO,皮层34.9±3.2 vs 53.1±1.7,P<0.05,纹状体54.0±3.0 vs 75.0±1.9;P<0.05,大脑半球34.7±2.1 vs 48.5±2.3,P<0.05,图4)。

图3 各组皮质、纹状体及总梗死面积比较Fig. 3 Comparison of the infarct size in cortex, striatum and total size among groups

图4 各组梗死面积比较Fig. 4 Comparison of the infarct sizes among saline+MCAO, IPC+saline+MCAO and IPC+Metformin+MCAO groups
3 讨 论
既往研究已证实IPC 72h后HSP70明显增多[19-20],故本研究选择这一时间点作为检测pAMPK的时间点。热休克反应是细胞在多种环境、生理压力下产生的一种很常见的保护作用[21],故选择HSP70作为产生缺血保护作用的标志。热休克反应中,热休克蛋白HSP表达明显增加[22],其中HSP70家族最常见于动物细胞[23]。有研究发现在热应激条件下,应用AMPK抑制剂可提高HSP70的表达,而应用AMPK激活剂AICAR则可抑制HSP70表达[24]。
本研究表明,IPC有明确的神经保护作用,且可先上调pAMPK而后下调pAMPK;给予AMPK抑制剂可显著减少梗死面积,但与IPC并无协同作用;给予AMPK激活剂二甲双胍可逆转IPC的神经保护作用。这些结果提示AMPK信号通路可能参与了脑缺血预处理的神经保护作用。
无论在脑或其他器官中,参与IPC保护作用的内源性机制都是目前的研究热点。早期实验证实5~10min的缺血处理会导致28%的脑组织出现显微镜可见的梗死[19],因此本研究选择3min短暂IPC以避免组织损伤。TTC(图1A)染色同样表明这种IPC未引起神经元损伤[15]。由于IPC后72h HSP70增多,且HSP70的保护作用已被证实[19-20],因此,本实验选择此时间点进行MCAO处理。
既往研究证实,外周组织器官(如心脏、肝脏等)中AMPK激活可引起缺血耐受,但在脑组织中AMPK水平增加却会加重缺血损伤[9,25]。脑内糖、氧不足时,神经细胞不能有效进行无氧糖酵解,AMPK在脑神经元中高表达时,能量储存下降的指标AMP/ATP比值即有所增高,激活AMPK,驱动ATP产生,导致新陈代谢进一步恶化[2]。缓慢下调脑内AMPK表达可保持缺血时ATP水平、减轻缺血诱导的乳酸酸中毒[9];而体外培养神经细胞中,短暂激活AMPK可增加葡萄糖载体3(GLUT3)表达,介导缺血耐受;慢性暴露于AMPK激活剂(如AICAR)可使AMPK超长时间激活或AMPK-α1过表达,均可导致神经细胞生存能力降低。AMPK调控的下游机制还可以通过激活延长因子-2激酶,调节IPC,抑制蛋白质在翻译过程中的延长,降低雷帕霉素靶蛋白(mTOn),减少蛋白质合成[4];AMPK也被认为是调节细胞自噬的重要介质,它可激活细胞自噬系统,参与缺血保护作用[26-27]。但是,目前AMPK诱导的相关分子信号通路仍有待明确。
AMPK活化状态会随时间的变化而改变。预处理之后早期(如4h),AMPK活化,pAMPK增加;在较晚时间点(如72h)进行MCAO处理时,pAMPK水平会下降,所以我们认为pAMPK的下降有利于迟发型IPC的神经保护作用。一些研究已应用AMPK激活剂二甲双胍对动物脑卒中急性或慢性治疗的效果进行测试,结果显示一次性大剂量给药后,动物脑内pAMPK水平增高,卒中所导致的代谢紊乱加重、乳酸产生增加、缺血损伤加剧[9];相反,在卒中处理前2周慢性小剂量给予二甲双胍,可抑制pAMPK的基础产生量和缺血诱导产生量,同时也出现神经保护作用。也有动物模型显示:药物激活AMPK会逆转IPC的神经保护作用[28]。本研究结果与这些结果相符,IPC后72h pAMPK水平下降、存在神经保护作用,AMPK活性降低表示代谢耐受。给予AMPK选择性抑制剂[17]复合物C后,实验动物脑内pAMPK水平明显降低[9,11,17,29],因此可通过抑制脑内AMPK活性而发挥神经保护作用。
本研究还证实,IPC可调控AMPK的表达,脑缺血时抑制AMPK活化有利于IPC的神经保护作用,但这种保护作用的反应时间和IPC后AMPK活化的下游信号通路仍有待进一步证实。我们认为,轻度、短暂的代谢应激可长期、缓慢地降低AMPK的活化,从而诱导代谢耐受。许多研究也表明寻找长期缓慢下调AMPK活性的方法可以保护缺血的脑组织,并尽早应用于临床急性脑缺血的治疗。
近年来,缺血耐受已成为脑缺血疾病临床治疗研究的新方向,其中远端缺血预处理方法已应用于部分院前急救。本研究提示AMPK信号途径参与了脑缺血预处理的神经保护作用,调控AMPK活性可能会诱导缺血耐受,有望成为临床缺血性卒中治疗的新靶点。
[1]Carling D, Clarke PR, Zammit VA, et al. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities[J]. Eur J Biochem, 1989, 186(1-2): 129-136.
[2]Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia[J]. J Cereb Blood Flow Metab, 2010, 30(3): 480-492.
[3]Hardie DG. Minireview: The AMP-activated protein kinase cascade: the key sensor of cellular energy status[J]. Endocrinology, 2003, 144(12): 5179-5183.
[4]Weisova P, Davila D, Tuffy LP, et al. Role of 5'-adenosine monophosphate-activated protein kinase in cell survival and death responses in neurons[J]. Antioxid Redox Signal, 2011, 14(10): 1863-1876.
[5]Gao P, Si LY, Xu Q, et al. Inhibitory effect of resveratrol on proliferation of vascular smooth muscle cells induced by angiotensin Ⅱ and its underlying mechanism[J]. Med J Chin PLA, 2013, 38(4): 269-273. [郜攀, 司良毅, 徐强, 等. 白藜芦醇对血管紧张素Ⅱ诱导的血管平滑肌细胞增殖的抑制作用及其机制观察[J]. 解放军医学杂志, 2013, 38(4): 269-273.]
[6]Wang Q, Yu CX, Gao L, et al. Thyroid-stimulating hormone regulates the phosphorylation of hepatic AMPKα Thr 172 instead of Ser 173[J]. J Shandong Univ (Health Sci), 2014, 52(6): 22-27. [王琦, 于春晓, 高聆, 等. 促甲状腺激素调节肝脏AMPKα Thr 172而非Ser 173的磷酸化[J]. 山东大学学报(医学版), 2014, 52(6): 22-27.]
[7]Hawley SA, Davison M, Woods A, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase[J]. J Biol Chem,1996, 271(44): 27879-2787.
[8]Bungard D, Fuerth BJ, Zeng PY, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation[J]. Science, 2010, 329(5996): 1201-1205.
[9]Li J, Benashski SE, Venna VR, et al. Effects of metformin in experimental stroke[J]. Stroke, 2010, 41(11): 2645-2652.
[10]Li J, Zeng Z, Viollet B, et al. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke[J]. Stroke, 2007, 38(11): 2992-2999.
[11]Stenzel-Poore MP, Stevens SL, King JS, et al. Preconditioning reprograms the response to ischemic injury and primes the em ergence of uniqueendogenous neuroprotective phenotypes: a speculative synthesis[J]. Stroke, 2007, 38(2 Suppl): 680-685.
[12]Yenari M, Kitagawa K, Lyden P, et al. Metabolic downregulation:a key to successful neuroprotection[J]? Stroke, 2008, 39(10): 2910-2917.
[13]Nishino Y, Miura T, Miki T, et al. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection[J]. Cardiovasc Res, 2004, 61(3): 610-619.
[14]Peralta C, Bartrons R, Serafin A, et al. Adenosine monophosphate-activated protein kinase mediates the protective effects of ischemic preconditioning on hepatic ischemiareperfusion injury in the rat[J]. Hepatology, 2001, 34(6): 1164-1173.
[15]Puisieux F, Deplanque D, Bulckaen H, et al. Brain ischemic preconditioning is abolished by antioxidant drugs but does not up-regulate superoxide dismutase and glutathion peroxidase[J]. Brain Res, 2004, 1027(1-2): 30-37.
[16]Deplanque D, Venna VR, Bordet R. Brain ischemia changes the long term response to antidepressant drugs in mice[J]. Behav Brain Res, 2011, 219(2): 367-372.
[17]McCullough LD, Zeng Z, Li H, et al. Pharmcaological inhibition of AMP-activated protein kinase provides neuroprotection in stroke[J]. J Biol Chem, 2005, 280(21): 20493-20502.
[18]Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action[J]. J Clin Invest, 2001, 108(8): 1167-1174.
[19]Zhan X, Kim C, Sharp FR. Very brief focal ischemia simulating transient ischemic attacks (TIAs) can injure brain and induce Hsp70 protein[J]. Brain Res, 2008, 1234: 183-197.
[20]Brown IR. Heat shock proteins and protection of the nervous system[J]. Ann N Y Acad Sci, 2007, 1113: 147-158.
[21]Dong L, Liu J, Ma HY, et al. Evaluation of immune effect of recombinant fusion protein targeting the prostate stem cell antigen based on PSCA and HSP70[J]. Med J Chin PLA, 2014, 39(9): 714-719. [董磊, 刘娟, 马红雨, 等. 重组蛋白PSCAHSP70的免疫活性及抗肿瘤效应观察[J]. 解放军医学杂志, 2014, 39(9): 714-719.]
[22]Hou YF, Bu PL. Change of serum heat shock protein 70 in the MODS patients and its clinical significance[J]. J Shandong Univ (Health Sci), 2013, 51(6): 64-70. [侯云峰, 卜培莉. 多器官功能障碍综合征患者血清热休克蛋白70的变化及其临床意义[J]. 山东大学学报(医学版), 2013, 51(6): 64-70.]
[23]Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism[J]. Cell Mol Life Sci, 2005, 62(6): 670-684.
[24]Wang T, Yu Q, Chen J, et al. PP2A mediated AMPK inhibition pr omotes HSP70 expression in heat shock response[J]. PLoS One, 2010, 5(10). pii: e13096.
[25]Gidday JM. Cerebral preconditioning and ischaemic tolerance[J]. Nat Rev Neurosci, 2006, 7(6): 437-448.
[26]Sheng R, Zhang LS, Han R, et al. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning[J]. Autophagy, 2010, 6(4): 482-494.
[27]Vingtdeux V, Chandakkar P, Zhao H, et al. Novel synthetic smallmolecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation[J]. FASEB J, 2011, 25(1): 219-231.
[28]Lotz C, Fisslthaler B, Redel A, et al. Activation of adenosinemonophosphate-activated protein kinase abolishes desfluraneinduced preconditioning against myocardial infarction in vivo[J]. J Cardiothorac Vasc Anesth, 2011, 25(1): 66-71.
[29]Liu F, Benashski SE, Persky R, et al. Age-related changes in AMP-activated protein kinase after stroke[J]. Age, 2012, 34(1): 157-168.
Effects of adenosine 5’monophosphate-activated protein kinase on europrotection induced by ischemic preconditioning
TIAN Yuan-ru-hua, ZHOU Zhong-he, CHEN Hui-sheng*
Department of Neurology, General Hospital of Shenyang Command, Shenyang 110840, China
*
, E-mail: chszh@aliyun.com
This work was supported by the Scientific and Technological Project of Liaoning Province(2013225089) and the National Natural Science Foundation of Liaoning Province(2013020204)
ObjectiveTo investigate the effects of adenosine 5'-monophosphate-activated protein kinase (AMPK) and phosphated AMPK (pAMPK) signals in ischemic preconditioning (IPC), and the effect of pharmacological intervention of AMPK on infarct size of the brain.MethodsA brief (3min) middle cerebral artery occlusion (MCAO) was employed to induce IPC in male rat, and another 90-min MCAO was performed 4 or 72h later. The levels of AMPK and pAMPK were assessed after IPC. A pharmacological activator metformin, or inhibitor compound C of AMPK, was used to analyze the correlation of IPC to AMPK signaling in MCAO rats.ResultsThe infarct size of total cerebral hemisphere and cortex was significantly decreased in MCAO animals by IPC for 72h (P<0.05, n=8), and the neurological deficit scores (NDS) of MCAO rats were also improved (P<0.05, n=8). There was a significant increase in pAMPK expression after a 90min MCAO (P<0.05, n=6), and a significant decrease in induced pAMPK expression (P<0.05, n=6) achieved only by a 72h IPC treatment. Intraperitoneal injection of an AMPK inhibitor, compound C, could decrease the infarct size in MCAO rats (P<0.05, n=6), but combined IPC (72h) and injection of compound C did not result in further decrease of the infarct size (P>0.05, n=6). The AMPK activator metformin can significantly reverse the protective effect of IPC (P<0.05, n=6).ConclusionsThe signals of AMPK and pAMPK play an important role in neuroprotective effect of IPC on cerebral ischemic injury. The neuroprotective effect of IPC may be associated with the down-regulation of pAMPK.
brain ischemia; infarction, middle cerebral artery; amp-activated protein kinases; metformin; compound C
R743.31
A
0577-7402(2015)05-0366-06
10.11855/j.issn.0577-7402.2015.05.07
2014-11-07;
2015-03-29)
(责任编辑:沈宁)
辽宁省科技攻关计划(2013225089);辽宁省自然科学基金(2013020204)
田园如画,硕士研究生。主要从事神经病学方面的研究
110840 沈阳 沈阳军区总医院神经内科(田园如画、周中和、陈会生)
陈会生,E-mail:chszh@aliyun.com
