基于cpDNA和rDNAITS片段分析西南地区青杨派杨树的系统发育与进化关系
2015-06-28李佳蔓周安佩何承忠
李佳蔓,员 涛,周安佩,纵 丹,李 旦,何承忠,4*
(1云南省高校林木遗传改良与繁育重点实验室,西南林业大学,昆明650224;2西南地区生物多样性保育国家林业局重点实验室,西南林业大学,昆明650224;3云南生物多样性研究院,西南林业大学,昆明650224;4西南山地森林资源保育与利用省部共建教育部重点实验室,西南林业大学,昆明650224)
基于cpDNA和rDNAITS片段分析西南地区青杨派杨树的系统发育与进化关系
李佳蔓1,2,员 涛1,2,周安佩1,2,纵 丹1,2,李 旦3,何承忠1,2,4*
(1云南省高校林木遗传改良与繁育重点实验室,西南林业大学,昆明650224;2西南地区生物多样性保育国家林业局重点实验室,西南林业大学,昆明650224;3云南生物多样性研究院,西南林业大学,昆明650224;4西南山地森林资源保育与利用省部共建教育部重点实验室,西南林业大学,昆明650224)
西南地区青杨派杨树种质资源丰富,可为杨树遗传改良提供珍贵的基因资源,但树种之间形态学差异细微,该研究以山杨作为外类群,测定了西南地区及其他地区杨属青杨派17个种(杂种)共36份样本的3个叶绿体片段(atpF-atpH、trnL-F和matK)和核糖体ITS片段,并对其进行系统发育分析,以探讨西南地区青杨派树种的系统进化关系。结果表明:(1)在所有样本中,3个叶绿体片段atpF-atpH、trnL-F、matK的长度分别为605~634bp、957~1 010bp、819bp,3个片段拼接后的联合序列包含29个变异位点和15个信息位点;ITS片段对齐后的长度为646bp,变异位点19个,信息位点17个。(2)基于叶绿体联合序列和ITS片段的杨属青杨派树种的平均遗传距离分别为0.001 3和0.003 6。叶绿体联合片段的MP和Bayes系统树树型基本一致,青杨派树种可以划分为2组,第1组由青杨、三脉青杨、大青杨和辽杨构成;第2组中的小叶杨、小青杨、川杨、德钦杨、昌都杨、乡城杨、康定杨、西南杨、滇杨和藏川杨不能有效区分,且均与缘毛杨的遗传关系较近。(3)基于ITS的系统树与叶绿体联合片段构建的系统树差异不大,仅在于第2组中的小青杨与小叶杨、川杨等差异明显,与第1组中的树种紧密聚拢。
西南地区;青杨派;叶绿体联合片段;ITS;系统发育
早在1905年,Dode首次就揭示了杨属(Populus L.)的系统发生关系和种间遗传分化,且至今依然是杨属植物分类学的基础,其系统发育模型包括三亚属六派[1]。经过一百多年的研究,现通常把杨属划分为5派,即白杨派(Leuce)、青杨派(Tacamahaca)、黑杨派(Aigeiros)、大叶杨派(Leucoides)和胡杨派(Turanga)[23]。青杨派是杨属中最大的一个派,中国拥有的青杨派树种最多,分布最广,其天然种的数量占中国杨树天然种的一半以上[4]。其中,西南地区(主要包括四川省、云南省和西藏东南部)为杨属青杨派树种分布的富集区之一,其基因资源丰富,特有种多,分布广,变异丰富,拥有大量的变种和天然杂种,这些树种蕴含了许多优良种质资源,为杨树遗传改良提供了珍贵的基因资源[5-9]。但由于青杨派树种派内种间极易进行自然杂交,产生了大量的自然杂交种,而大树上的短枝叶变异大,使其表型多样性丰富,造成了系统分类上很大的困难[10]。另外,西南地区青杨派树种大部分处于野生状态,其开发和利用尚未受到研究者及公众的普遍认识,导致其基因资源在杨树遗传改良上应用较少。因此,开展杨树青杨派树种的系统发育关系研究,不仅有助于杨属青杨派系统分类,也能够为杨属青杨派树种资源的开发和利用提供前期基础。
分子生物技术的兴起,为植物系统发育的研究开辟了新的路径,使其在研究方法和策略上均发生了变革[11]。在高等植物基因组中,线粒体基因组进化速率较慢,遗传分化较小,不适合用于系统发育分析[1213],故在对植物的系统发育研究中,通常从核基因组和叶绿体基因组中寻找合适的片段。李岩等[14]以3株岩荞菜属(Aethionema L.)植株作为外类群,结合核糖体ITS测序结果和GenBank数据库中相应数据分析了同科菘蓝属(Isatis L.)宽翅菘蓝(I.violascens)27个样的系统关系和分类地位,通过构建的最大简约树(Maximum parsimony tree,MP tree)、最大似然树(Maximum likelihood tree,ML tree)和贝叶斯树(Bayesian tree)将样本聚分为2个分支,支持将宽翅菘蓝与I.emarginata合并为一个物种。高丽霞等[15]通过对27个样地的10种棘豆属(Oxytropis DC.)植物的54个单株的种间关系和系统进化分析,表明trnL-F序列可用于棘豆属种间系统发育关系的研究。狄红艳等[16]以白花草木樨(Melilotus alba)和黄花草木樨(Melilotus officinalis)的18个地理种群为材料,分析了其ITS和trnL-F序列。Gouja等[17]利用ITS、trnL-F和rbcL评价了突尼斯沙漠中沙拐枣属(Calligonum L.)3个待定种的31份个体间的系统关系,3个待定种在3片段联合后的分支图中均表现出良好的单系性。
单基因片段序列能提供的信息量有限,很多研究已不再满足于用单一片段序列来进行系统发育的研究,如Soltis等[18]利用来自叶绿体和核基因组的3个基因片段对被子植物进行系统发育的研究;Bayly等[19]比较了桉树3个属(Eucalyptus,Corymbia,Angophora)39个树种叶绿体基因组的结构特点,并通过构建MP树和Bayes树区分了桉树的主要分支,同时证明了叶绿体基因组变异片段能应用于低水平遗传距离物种间的研究。这种采用多基因序列进行系统发育重建的方法逐渐被广泛采用,成为分子系统发育研究的一种基本方法[20-22]。
目前对西南地区乡土杨树的分类学研究均是基于表型特征而来,从分子生物学水平获得的依据相对较少,仅陈珂[23]和Wan等[24]对其系统关系进行了探讨。基于此,本研究以西南地区杨属青杨派树种为主体材料,4个北方青杨派树种和1份小叶杨材料为补充材料,选取其叶绿体atpF-atpH、trnLF和matK等3个片段及核糖体ITS片段,以探讨西南地区杨属青杨派树种间的系统发育关系,为西南地区杨属青杨派树种的系统分类和遗传改良提供科学依据。

表1 杨树样本采集地Table 1 The sample sites of Populus
1 材料和方法
1.1 材 料
杨属青杨派树种于2012年9月采集于云南省西北部(香格里拉县、德钦县、维西县)、四川省甘孜州(乡城县、稻城县、理塘县、雅江县、康定县、汉源县)和西藏昌都地区(芒康县、昌都县、贡觉县),1份小叶杨样本来自于北京,小青杨、青杨和辽杨来自于内蒙古,大青杨采自于吉林,包括青杨派17个种/杂种以及作为外类群的白杨派山杨,共计36份样本(表1)。鉴于本研究主要研究种间系统发育关系,按照分层抽样理论,尽可能在种水平上较多选取材料,而种内只随机选取单株2~3株。采集杨树各样株幼嫩叶片,变色硅胶干燥保存,带回实验室备用。
1.2 实验方法
1.2.1 PCR扩增与测序 硅胶干燥后的样株叶片经冷冻混合球磨仪研磨后,采用SDS法并稍加改良,提取基因组总DNA,用0.6%琼脂糖凝胶电泳和核酸检测仪共同检测DNA的浓度和纯度,稀释至所需浓度(50~100ng/μL)后于-20℃冰箱中保存。
选取在物种系统发育分析中应用较广的叶绿体atpF-atpH、trnL-F和matK3个片段,及核糖体ITS片段进行扩增,引物序列及来源见表2。扩增反应总体系为25μL,其中正反引物各1μL(浓度为5pmol),DNA模板1μL,2×Taq MasterMix 12.5 μL,去离子水9.5μL。PCR反应条件:94℃预变性4min;94℃变性30s,退火45s(退火温度因引物而异),72℃延伸1min,共35个循环;72℃总延伸10 min。扩增产物送往北京华大基因科技股份有限公司进行纯化和测序。
1.2.2 数据分析 所得序列采用Bioedit 7.09软件[29]进行序列的拼接和人工校对,使用Clustal X 1.8[30]进行多序列的对比,利用MEGA5.02[31]对序列的长度、碱基组成和位点特性进行分析,通过Kimura 2-parameter(K2P)模型计算种间遗传距离。
用PAUP4b10软件[32]对36份供试材料的aptF-atpH、trnL-F、matK、ITS片段进行非一致性长度检验(ILD检验),只有当P>0.05时才能对序列进行合并。采用PAUP软件的MP法和Mrbayes 3.1.2软件的Bayes法构建系统发育树,其中,MP采用1 000次bootstrap循环[33]检验各分支的支持率,并计算树长(Length)、总一致性指数(CI)、总存留指数(RI)和尺度化后的一致性指数(RC);Bayes的模型选择是通过Modeltest 3.7[34]比较分析,以AIC为准则进行筛选确定,采用马儿科夫链的蒙特卡洛法(Markov chain Monte Carlo Process),以随机树为起始,运行1 000 000代,每100代抽样1次,舍弃2 500个老化样本(占25%)后,以剩余样本构建一致树。
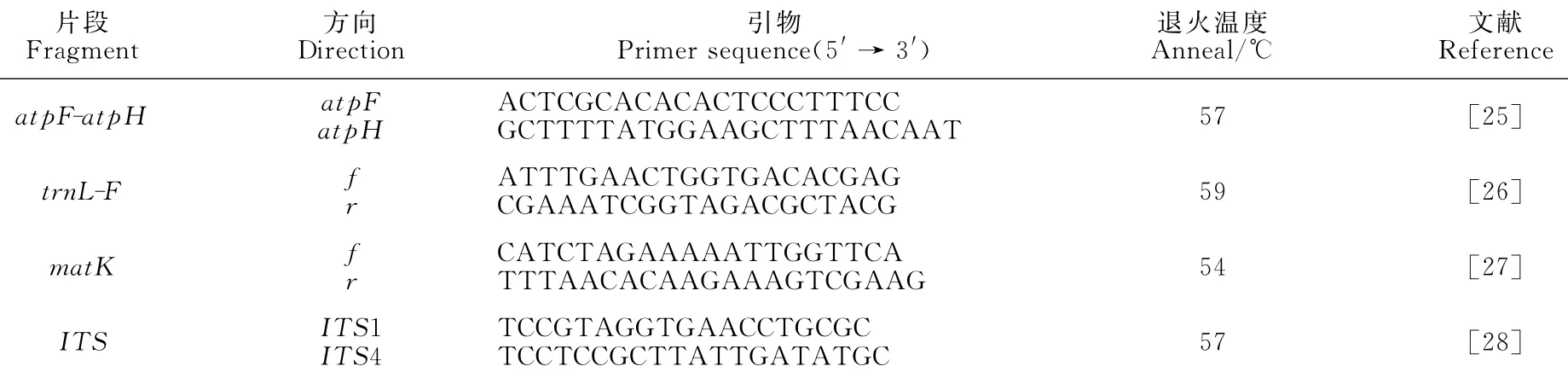
表2 扩增引物及来源Table 2 PCR primers and sources used in this study
2 结果与分析
2.1 基因序列的差异分析
由表3可看出,36份杨树材料的atpF-atpH、trnL-F和matK片段的平均长度分别为629(605~634)、995(957~1010)和819bp,其中trnL-F片段序列包含的变异位点和信息位点数最多,分别为12个和8个。3个叶绿体片段联合后的长度为2 381~2 463bp,平均长度为2 443bp,其中,变异位点29个,信息位点15个,G+C含量为31.6%。ITS片段的长度范围为635~645bp,平均长度为642 bp,含有变异位点19个,信息位点17个,G+C含量为65.5%。
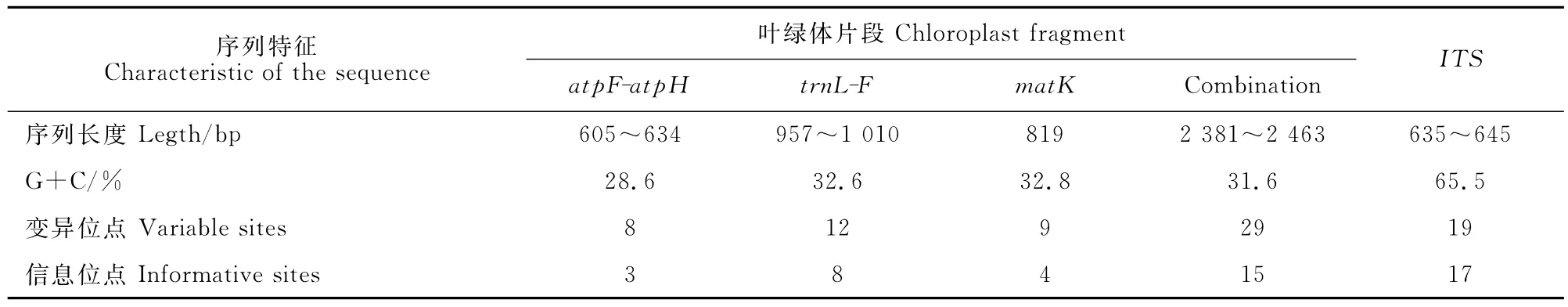
表3 atpF-atpH、trnL-F、matK、ITS片段的序列特征Table 3 Characteristics of the atpF-atpH,trnL-F,matKand ITSsequences
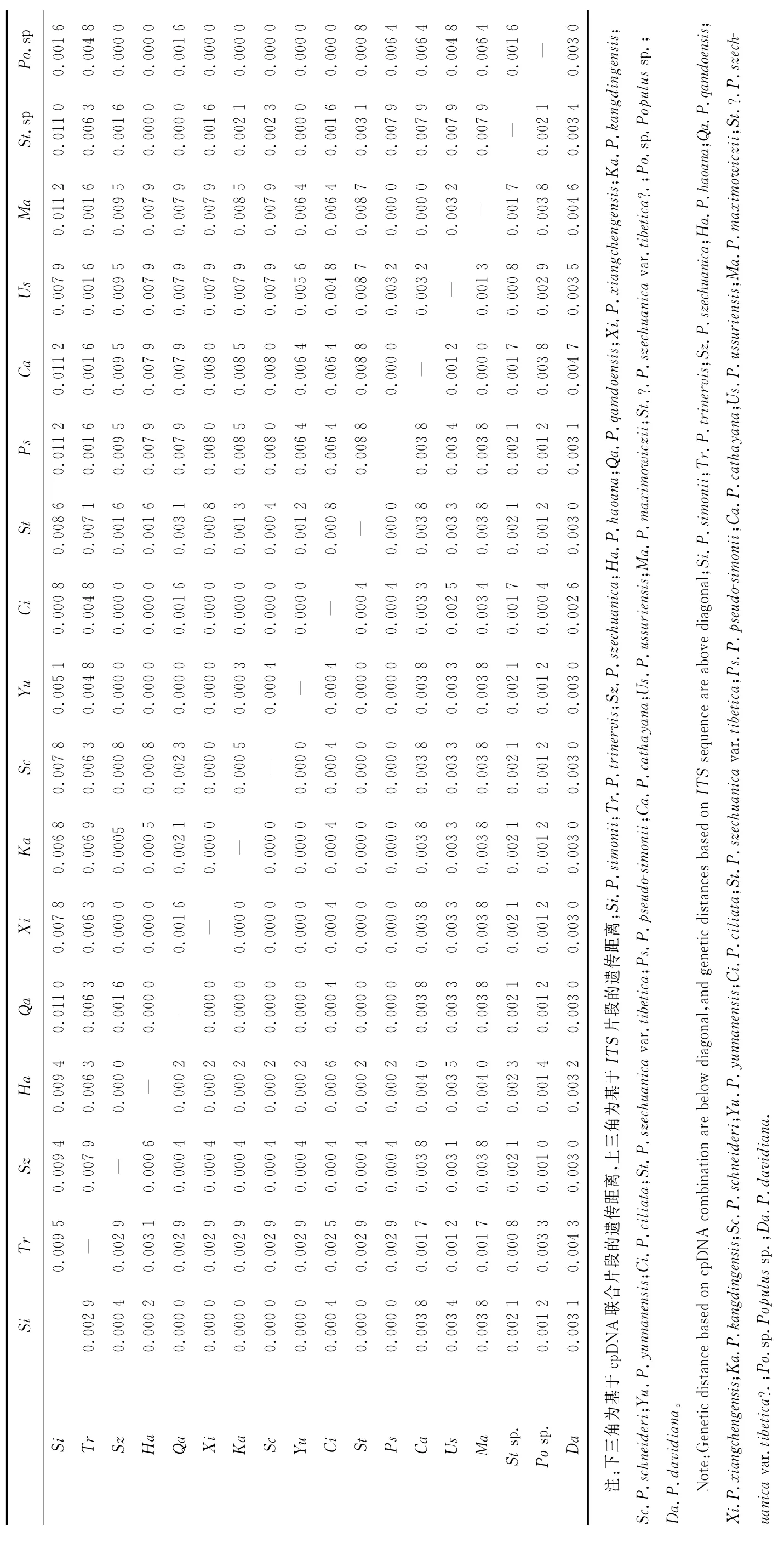
表4 青杨派树种间的遗传距离Table4 GeneticdistancesamongspeciesinsectionTacamahaca
2.2 杨树青杨派树种的遗传距离分析
以青杨派17个种和杂种及外类群山杨的cpDNA及ITS序列做为数据分析的基础,采用MEGA软件的K2P模型计算树种之间的遗传距离(表4)。基于叶绿体联合片段的遗传距离表明,除去疑似种和外类群,青杨派树种的平均遗传距离较小,为0.001 3,可分为2组。三脉青杨、青杨、大青杨和辽杨4个树种共同组成第1组,组内树种的遗传距离在0.0000~0.001 7之间;剩余树种为第2组,组内遗传距离较小(0.0000~0.000 6);2个组树种间的遗传距离在0.002 5~0.004 0之间。ITS序列间的平均遗传距离为0.003 6,同样可分为2组,分组情况与叶绿体联合片段类似,第1组仅多了小青杨,组内遗传距离较小(0.0000~0.003 2),第2组(不含小青杨)的组内遗传距离较大(0.0000~0.011 0)。
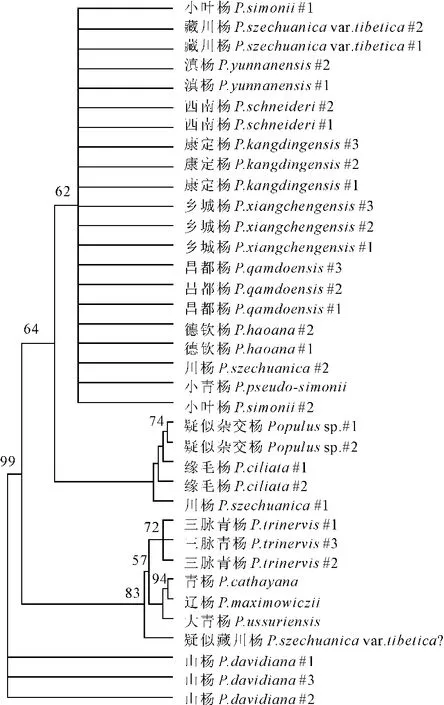
图1 基于atpF-atpH、trnL-F、matK片段杨属青杨派树种的最大简约树节点处仅显示超过50%的置信值(Bootstrap支持率)Fig.1 Maximum parsimony tree in sect.Tacamahacaof Populus based on atpF-atpH,trnL-Fand matK Confidence values(Bootstrap support values)are reported for nodes over 50%
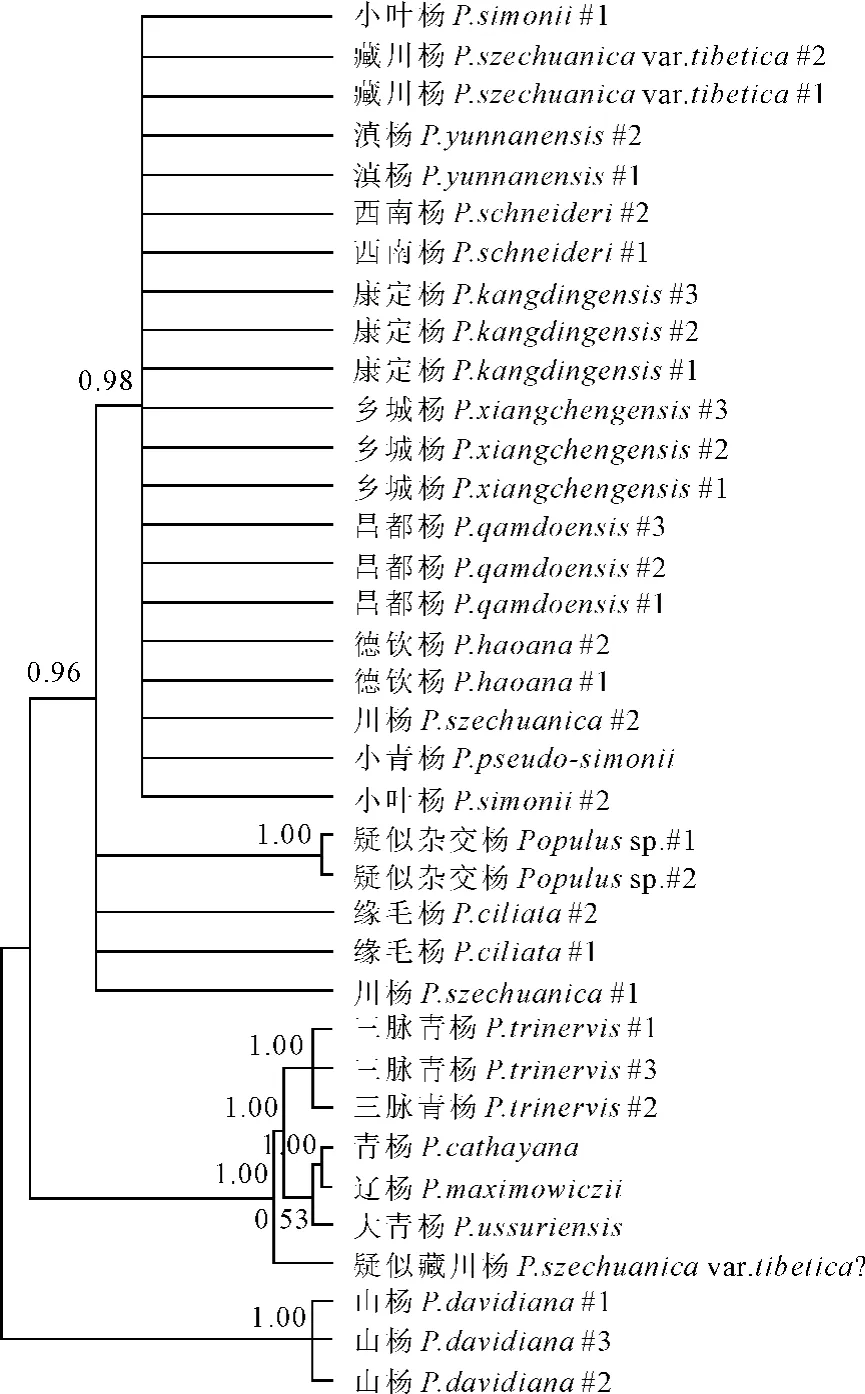
图2 基于atpF-atpH、trnL-F、matK片段杨属青杨派树种的贝叶斯树节点处仅显示超过50%的置信值(后验概率)Fig.2 Bayesian tree in sect.Tacamahacaof Populus based on atpF-atpH,trnL-Fand matK Confidence values(posterior probability values)are reported for nodes over 50%
2.3 杨属青杨派树种的系统发育分析
单独分析叶绿体atpF-atpH、trnL-F和matK片段的系统发育树,其支持率和分辨率均较低,通过ILD检测后发现,3种片段具有极高的同质性(P=1.00>0.05),因此,合并后的联合片段可以用于系统发育分析。其中,MP树的一致性指数(CI)为0.909 1,保持性指数(RI)为0.969 2,存留一致性指数(RC)为0.881 1;Bayes树的最佳进化模型为F81+I。用两种方法得到的系统树树型相似(图1和图2),均先分离作为外类群的3株山杨,然后将青杨派树种分为2个分支,第1分支包括青杨、辽杨、大青杨、三脉青杨和疑似藏川杨的天然杂种,其置信值达83%(MP树)和100%(Bayes树),其中,青杨和辽杨具有极高的相似性,MP和Bayes的置信值分别为94%和100%;其余26株青杨派树种构成第2分支(MP树和Bayes树的置信值分别为64%和96%),其中,缘毛杨、疑似杂交种和1株川杨个体位于第2分支的基部,而小叶杨、滇杨、小青杨、川杨(1个个体)、藏川杨、乡城杨、康定杨、西南杨、昌都杨和德钦杨则以姐妹种的形式聚类,其分支互为平行支,表明这10个种之间的系统关系不够明确。
基于ITS片段的MP树(CI=0.863 6,RI=0.958 3,RC=0.827 7)和Bayes树(最佳进化模型为TrN+I)的分析表明,两种系统树均表明所有青杨派树种聚在一起,与外类群山杨明显分离,而青杨派树种也分为2个分支,其中辽杨、青杨、小青杨、大青杨和三脉青杨为第1分支,位于系统树的基部,MP树和Bayes树的置信值分别为53%和71%。其余的树种构成第2分支,位于系统树的顶部,其MP树和Bayes树的置信值分别为50%和96%。在第2分支中,MP树(图3)和Bayes树(图4)的树型有一定的差异,其中MP树首先分出2株小叶杨,最后分出3株昌都杨和1株疑似藏川杨的天然杂种,而Bayes树中,2株小叶杨个体以73%的置信值聚类后,再与其他杨树个体呈姐妹种关系相聚。由此可见,基于ITS序列依然不能够明确第2分支树种之间的系统关系。


图4 基于ITS片段杨属青杨派树种的贝叶斯树节点处仅显示超过50%的置信值(后验概率)Fig.4 Bayesian tree in sect.Tacamahacaof Populus based on ITS Confidence values(posterior probability values)are reported for nodes over 50%
3 讨 论
3.1 青杨派杨树cpDNA和ITS片段的序列特征
高等植物细胞中具有3个基因组,即核基因组、叶绿体基因组和线粒体基因组,由于陆地植物的线粒体基因进化速率相对较慢,并不适合用于系统发育的研究,故而对植物的研究主要集中于核基因组和叶绿体基因组[35-37]。卫尊征等[38]对杨属4派17个主要栽培种及杂种共计26个单株的trnL-F间隔区序列进行了研究,其结果显示,26个单株基因trnL-F间隔区序列的长度为771~811bp,对齐后长度为857bp,具有46个变异位点,33个信息位点。而本研究中,trnL-F片段的长度为957~1 010 bp,对齐后为995bp,变异位点12个,信息位点8个,与前者所报道的结构具有一定的差异,主要原因可能为取材不同(前者包含杨属4个派,本研究仅为青杨派树种)、外类群(前者为同科不同属的蒿柳和极地柳,本研究为同属不同派的山杨)和序列片段范围(前者根据序列自行设计800bp左右的引物,本研究采用Taberlet开发的通用引物)的选择不同。
ITS为核糖体DNA(nrDNA)的转录间隔区序列片段,由于其进化速率快而被广泛应用于属和属以下分类阶元的系统发育研究中[28,39-41]。如Abdollahzadeh等[42]以杨树2个种为外类群,构建了柳树55个种62个样ITS片段的MP树和Bayes树。Yost等[43]利用ITS片段分析景天科(Crassulaceae)仙女杯属(Dudleya)二倍体物种的系统发育和三倍体物种的进化起源,结果表明二倍体物种虽可分成4个分支,但很多种间关系尚不能确定,三倍体物种ITS片段上的信息量也不足以解决其起源问题。在杨树的ITS研究中,史全良等[44]以旱柳和簸箕柳为外类群,对杨属5派15个代表种的ITS片段进行了分析,其结果表明,ITS1和ITS2长度分别为220~223bp与201~210bp,5.8S为164 bp,总长度为594bp,共有92个变异位点和61个信息位点,其中ITS1中变异位点39个,信息位点17个,ITS2的变异位点和信息位点分别为48个和44个。而在本研究中,ITS片段的序列长度为635~645bp,平均为642bp,对齐后为646bp,变异位点只有19个,信息位点为17个,与前者的报道有一定的差异,主要也是由于研究材料、外类群选择以及序列片段范围选择的不同造成的。
3.2 西南地区青杨派树种系统进化关系
中国西南地区既有高原平面又有高山峡谷,地形地貌复杂,立体气候和环境多样,冰川活动,人文地史条件独特,特别是横断山区以山川骈列,南北走向,在第四纪冰期来临时,为植物的退避提供了优异的条件,为主要分布于北半球温带和寒带的杨树退避提供了通道[5]。因此,使该区孕育了丰富的生物资源,杨树种类之多,性状变异之丰富,是世界其他地方少有的[7]。然而,据现有资料报道,西南地区分布的青杨派乡土杨树既有广布树种,如小叶杨、青杨、滇杨、川杨、藏川杨等,也有地域性较强的窄布树种(或称为地方小种),如德钦杨、康定杨、乡城杨、西南杨、三脉青杨、昌都杨等,且其形态特征一般与广布树种十分相似,仅存细微差异,如西南杨与青杨相似,仅是西南杨的枝、叶、果序和果均密被毛;乡城杨与川杨相似,仅是乡城杨的小枝、叶柄、叶脉和果序轴均被毛[2,6,45]。
不可否认的是,形态特征的差异是经典分类学中划分植物不同种类的重要依据,而微小的差异也可能是同一物种在异质环境条件中长期进化的适应性表现。尤其是中国西南地区,物种分布区域的山脉阻隔特点突出,地势高峻,冰川作用强烈[46]。而冰川作用更是促进植物演化的主要动力,每次冰期气候来临,植物在寒冷气候作用下必然不断地产生适应性,以新的形态、结构和习性适应新的环境。由此引出的问题是,当前西南地区分布的特有杨树种,是否由于气候条件的变化、因地理隔离和地域间气候条件的差异而由原来的广布树种在形态特征等方面产生适应性?赵能等[8]依据表型性状将西南杨归为康定杨的变种。而Wan等[24]利用AFLP标记和叶绿体trnT-F片段对西南地区38份杨树材料的亲缘关系进行了分析,结果表明西南杨可能并非康定杨的变种,而是康定杨和青杨的自然杂交种。本研究对西南地区杨属青杨派树种的研究结果表明,基于叶绿体基因组atpF-atpH、trnL-F和matK联合片段的MP和Bayes系统树基本一致,小叶杨、小青杨、川杨、德钦杨、昌都杨、乡城杨、康定杨、西南杨、滇杨、藏川杨以姐妹种的形式共同聚为一个分支,之间的系统关系不能确定。他们与缘毛杨的遗传关系较近,但却与青杨、三脉青杨、大青杨、辽杨所构成的分支遗传关系较远。基于ITS的MP和Bayes系统树与叶绿体基因组联合片段的系统树差异不大,其主要不同在于小青杨与青杨、辽杨等杨树紧密聚在一起,表明小青杨很可能为青杨派2个分支树种所产生的古老自然杂交种。
通过比较形态学特征和系统进化关系发现,有些形态特征相似的树种在系统树中紧密聚类,如乡城杨、德钦杨、昌都杨、康定杨、藏川杨、川杨、滇杨等,但地方小种如德钦杨、康定杨、乡城杨等是否为广布树种小叶杨、川杨、藏川杨、滇杨等的一种环境适应型尚不能确定,有待进一步研究。此外,青杨与川杨、滇杨、西南杨等树种之间,三脉青杨和小叶杨之间的形态特征相近,但从遗传关系来看,无论是基于叶绿体基因组、还是核基因组ITS片段,都表明他们之间具有明显的差异,这很可能是杨属派内种间频繁地自然杂交现象所导致。
综上所述,西南地区青杨派乡土树种之间的遗传关系较为复杂,虽然赵能等[6-9]经过长期研究,基于形态特征建立了西南地区乡土杨树的分类系统,但从分子生物学方面开展的研究却较少。本研究基于叶绿体基因组3个片段的联合和核基因组ITS序列分析了该区域青杨派树种的系统进化关系,初步将西南地区青杨派树种归为2个分支,第1分支为采集于该地区的小叶杨、滇杨、川杨、藏川杨、乡城杨、康定杨、西南杨、昌都杨、德钦杨,第2分支为三脉青杨,但每个分支内的树种系统关系仍不能够明确,需要借助更多的分子生物学技术深入地开展研究。
[1] 斯塔罗娃.杨柳科的育种[M].马常耕,译.北京:科学技术文献出版社,1984:1-31.
[2] 王 战,方振富.中国植物志(第20卷,第2分册)[M].北京:科学出版社,1984:2-78.
[3] 徐纬英.杨树[M].哈尔滨:黑龙江人民出版社,1988:13-15.
[4] LUAN H H(栾鹖慧),SU X H(苏晓华),ZHANG B Y(张冰玉).Research progress in genetic evaluation of Populus L.germplasm resources[J].Chinese Bulletin of Botany(植物学报),2011,46(5):586-595(in Chinese).
[5] GONG G T(龚固堂).The geographic distribution and origin of Populus L.[J].Journal of Sichuan Forestry Science and Technology(四川林业科技),2004,25(2):25-30(in Chinese).
[6] ZHAO N(赵 能).Taxonomic study on Salicaceae in Sichuan and ITSadjacent regions(Ⅲ)[J].Sichuan Forestry Science and Technology(四川林业科技),1994,15(2):1-11(in Chinese).
[7] YU SH Q(余树全),LIU J(刘 军),FU D R(付达荣),et al.Characteristics of Tacamahaca genes in the Western Sichuan Plateau[J].Journal of Zhejiang Forestry College(浙江林学院学报),2003,20(1):27-31(in Chinese).
[8] ZHAO N(赵 能),LIU J(刘 军).Taxonomic studies on Populus L.in Southwestern China(Ⅰ)[J].Journal of Wuhan Botanical Research(武汉植物学研究),1991,9(3):229-238(in Chinese).
[9] LIU J(刘 军).Geographical condition of section Tacamahacain the Western Sichuan Plateau[J].Sichuan Forestry Science and Technology(四川林业科技),1997,18(2):36-39(in Chinese).
[10] YIN CH Y(尹春英),PENG Y H(彭幼红),LUO J X(罗建勋),et al.Advances in research on genetic diversity in Populus[J].Acta Phytoecologica Sinica(植物生态学报),2004,28(5):711-722(in Chinese).
[11] 邹喻苹,葛 颂,王晓东.系统与进化植物学中的分子标记[M].北京:科学出版社,2001:159-165.
[12] PENNISI E.Wanted:A barcode for plants[J].Science,2007,318:190-191.
[13] KRESS W J,ERICKSON D L.DNA barcodes:Genes,genomics,and bioinformatics[J].Proceedings of National Academy of Sciences of the United States America,2008,105(8):2 761-2 762.
[14] LI Y(李 岩),QI A(齐阿尔达尔),ZHANG ZH F(张志峰),et al.Phylogenetic relationship and taxonomic status of Isatis violascens Bunge(Isatis,Brassicaceae)[J].Acta Bot.Boreal.-Occident.Sin.(西北植物学报),2014,33(5):902-907(in Chinese).
[15] GAO L X(高丽霞),LU P(卢 萍),JIN F(金 凤),et al.trnL-Fsequences analysis and molecular phylogeny of 10species of Oxytropis[J].Acta Bot.Boreal.-Occident.Sin.(西北植物学报),2013,33(2):266-271(in Chinese).
[16] DI H Y(狄红艳),LUO K(骆 凯),ZHANG J Y(张吉宇),et al.Genetic diversity analysis of Melilotus populations based on ITSand trnL-trnFsequences[J].Acta Bot.Boreal.-Occident.Sin.(西北植物学报),2014,34(2):265-269(in Chinese).
[17] GOUJA H,FERNANDEZ A G,GARNAJE T,et al.Genome size and phylogenetic relationships between the Tunisian species of the genus Calligonum(Polygonaceae)[J].Turkish Journal of Botany,2014,38(1):13-21.
[18] SOLTIS P S,SOLTIS D E,CHASE M W.Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology[J].Nature,1999,402:402-404.
[19] BAYLY M J,RIGAULT P,SPOKEVICIUS A,et al.Chloroplast genome analysis of Australian eucalypts-Eucalyptus,Corymbia,Angophora,Allosyncarpiaand Stockwellia(Myrtaceae)[J].Molecular Phylogenetics and Evolution,2013,69(3):704-716.
[20] WENDEL J F,DOYLE J J.Phylogenetic incongruence:window into genome history and molecular evolution[M]//SOLTIS D E,SOLTIS
P S,DOYLE J J.Molecular Systematics of Plants II:DNA Sequencing.Boston:Kluwer Academic Publishers,1998:265-296.
[21] CRAWFORD D J.Plant macromolecular systematics in the past 50years:one view[J].Taxon,2000,49(3):479-501.
[22] DELSUC F,BRINKMANN H,PHILIPPE H.Phylogenomics and the reconstruction of the tree of life[J].Nature Reviews Genetics,2005,6(5):361-375.
[23] 陈珂.川西青杨组(Section Tacamahaca Spach)不同种的亲缘关系分析[D].成都:中国林科院成都生物研究所,2007.
[24] WAN X Q,ZHANG F,ZHONG Y,et al.Study of genetic relationships and phylogeny of the native Populusin Southwest China based on nucleotide sequences of chloroplast trnT-trnFand nuclear DNA[J].Plant Systematics and Evolution,2013,299(1):57-65.
[25] LAHAYE R,SAVOLAINEN V,DUTHOIT S,et al.A test of psbK-psbI and atpF-aptHas potential plant DNA barcodes using the flora of the Kruger National Park as a model system(South Africa)[J].Available from Nature Proceedings,http://hdl.handle.net/lolol/npre.12008.11896.10101.
[26] TABERLET P,GIELLY L,PAUTOU G,et al.Universal primers for amplification of three non-coding regions of chloroplast DNA[J].Plant Molecular Biology,1991,17(5):1 105-1 109.
[27] VON CRAUTLEIN M,KORPELAINEN H,PIETILAINEN M,et al.DNA barcoding:a tool for improved taxom identification and detection of species diversity[J].Biodiversitty Conservation,2011,20(2):373-389.
[28] WHITE T J,BRUNS T,LEE S,et al.Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics[M]//INNIS M A,GELFAND D H,SNINSKY J J.PCR Protocols:a Guide to Methods and Applications.San Diego:Academic Press,1990:315-322.
[29] HALL T.BioEdit:a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT[J].Nucleic Acids Symposium Series,1999,41:95-98.
[30] THOMPSON J D,GIBSON T J,PLEWNIAK F,et al.CLUSTAL-X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Research,1997,25(24):4 876-4 882.
[31] TAMURA K,PETERSON D,PETERSON N,et al.MEGA5:Molecular evolutionary genetics analysis using maximum likelihood,evolutionary distance,and maximum parsimony methods[J].Molecular Biology and Evolution,2011,28(10):2 731-2 739.
[32] SWOFFORD D L.PAUP*4.0:Phylogenetic analysis using parsimony(*and other methods)[M].Sunderland:Sinauer Associates,2001.
[33] FELSENSTEIN J.Phylogenies from molecular sequences:Inference and reliability[J].Annual Review of Genetics,1988,22(1):521-565.
[34] POSADA D,CRANDALL K A.Modeltest:testing the model of DNA substitution[J].Bioinformatics,1998,14(9):817-818.
[35] REN B Q(任保青),CHEN ZH R(陈之端).DNA barcoding plant life[J].Chinese Bulletin of Botany(植物学报),2010,45(1):1-12(in Chinese).
[36] LI X W,YANG Y,HENRY R J,et al.Plant DNA barcoding:from gene to genome[J].Biological Reviews,2015,90(1):157-166.
[37] AHMED I,MATTHEWS P J,BIGGS P J,et al.Identification of chloroplast genome loci suitable for high-resolution phylogeographic studies of Colocasia esculenta(L.)Schott(Araceae)and closely related taxa[J].Molecular Ecology Resources,2013,13(5):929-937.
[38] WEI Z ZH(卫尊征),GUO L Q(郭丽琴),ZHANG J F(张金凤),et al.Phylogenetic relationship of Populus by trnL-Fsequence analysis[J].Journal of Beijing Forestry University(北京林业大学学报),2010,32(2):27-33(in Chinese).
[39] GAO L M(高连明),LI D ZH(李德铢).Phylogenetic relationships of Rhododendronsection Azaleastrum(Ericaceae)based on ITSsequences[J].Acta Phytotaxonomica Sinica(植物分类学报),2003,41(2):173-179(in Chinese).
[40] LI Z M,LI J,LI X W.Polyphyly of the genus Actinodaphne(Lauracea)inferred from the analyses of nrDNAITSand ETSsequences[J].Acta Phytotaxonomica Sinica,2006,44(3):272-285.
[41] WANG Q,RICHARD J A,YU Q S,et al.Pleistocene climate change and the origin of two desert plant species,Pugionium cornutumand Pugionium dolabratum(Brassicaceae),in Northwest China[J].New Phytologist,2013,199(1):277-287.
[42] ABDOLLAHZADEH A,KAZEMPOUR O S,MAASSOUMI A A.Molecular phylogeny of the genus Salix(Salicaceae)with an emphasize to ITSspecies in Iran[J].The Iranian Journal of Botany,2011,17(2):244-253.
[43] YOST J M,BONTRAGER M,MCCABE S W,et al.Phylogenetic relationships and evolution in Dudleya(Crassulaceae)[J].Systematic Botany,2013,38(4):1 096-1 104.
[44] SHI Q L(史全良),ZHUGE Q(诸葛强),HUANG M R(黄敏仁),et al.Phylogenetic relationship of Populus sections by ITSsequence analysis[J].Acta Botanica Sinica(植物学报),2001,43(3):323-325(in Chinese).
[45] 《四川植物志》编辑委员会.四川植物志(第3卷)[M].成都:四川科学技术出版社,1985:39-58.
[46] SUN D Q(孙殿卿),ZHOU M L(周慕林),PAN J Y(潘建英).Quaternary glaciations in China[J].Acta Genologica Sinica(地质学报),1977,51(2):101-107(in Chinese).
(编辑:宋亚珍)
Phylogeny of Poplar in Section TacamahacaSpecies from Southwest China Based on Sequence Data of cpDNA Fragments and rDNAITS
LI Jiaman1,2,YUN Tao1,2,ZHOU Anpei1,2,ZONG Dan1,2,LI Dan3,HE Chengzhong1,2,4*
(1Key Laboratory for Forest Genetic and Tree Improvement &Propagation in Universities of Yunnan Province,Southwest Forestry University,Kunming 650224,China;2Key Laboratory of Biodiversity Conservation in Southwest China,State Forestry Administration,Southwest Forestry University,Kunming 650224,China;3Yunnan Academy of Biodiversity,Southwest Forestry U-niversity,Kunming 650224,China;4Key Laboratory for Forest Resources Conservation and Use in the Southwest Mountains of China,Ministry of Education,Southwest Forestry University,Kunming 650224,China)
Despite abundant germplasm resources in Southwest China for poplar genetic improvement,the varied species exist tiny difference in morphology and have unknown systematic relationship.Using Populus davidianaas outgroup,the phylogenetic relationship of 36specimens covering 17species or hybrids collected from Southwest China and other areas was explored based on sequence data of chloroplast atpF-atpH,trnL-F,matKand nuclear ITS.The results showed that:(1)the ranges of length in atpF-atpH,trnL-Fand matKwere 605-634bp,957-1 010bp and 819bp respectively,and the combination of 3chloroplast fragments had 29variable sites and 15informative sites,while the aligned length of ITSfragment including 19variable sites and 17informative sites was 646bp.(2)The average genetic distance among all samples of chloroplast combination was 0.001 3and that of ITSfragment was 0.003 6.Based on chloroplast combination data,the result of MP tree was consistent with that of Bayesian algorithms and both suggested the Populus section Tacamahacaspecies were divided into 2clades,of which,clade 1was formed by P.cathayana,P.trinervis,P.ussuriensis,P.maximowiczii,and the species in clade 2,namely,P.simonii,P.pseudosimonii,P.szechuanica,P.haoana,P.qamdoensis,P.xiangchengensis,P.kangdingensis,P.schneideri,P.yunnanensis,P.szechuanicavar.tibetica,could not be clearly separated and had close relationship with P.ciliata.(3)Although a few differences in phylogenetic tree based on chloroplast combination data,ITS fragment provided that P.pseudo-simonii existed closer affinity with other species in clade 1(e.g.P.simonii and P.szechuanica)but larger genetic distance with others in clade 2.The results of this study made a understand on the phylogeny of poplar in section Tacamahacaspecies from Southwest China,and provided a scientific bases for their systematic classification and evolutionary relationship.
Southwest China;Tacamahaca;chloroplast combination;ITS;phylogeny
Q789
A
10.7606/j.issn.1000-4025.2015.06.1113
1000-4025(2015)06-1113-10
2015-02-06;修改稿收到日期:2015-05-17
国家林业公益性行业专项(201104076);国家自然科学基金(31360184,31460205);云南省教育厅基金(2014J099);云南省中青年学术与技术带头人后备人才培养基金(2012HB021)
李佳蔓(1989-),女,在读硕士研究生,主要从事林木遗传育种研究。E-mail:lijiaman16@126.com
*通信作者:何承忠,博士,教授,主要从事林木遗传育种与分子生物学研究。E-mail:hcz70@163.com
