高效液相色谱法测定鸡肌肉中地克珠利、妥曲珠利及其代谢物的残留量
2015-06-13林仙军陆春波包爱情周芷锦罗成江李孝军应永飞
林仙军,陆春波,包爱情,周芷锦,罗成江,李孝军,应永飞∗
(1.浙江省兽药饲料监察所,杭州311000;2.舟山出入境检验检疫局,浙江舟山316000)
高效液相色谱法测定鸡肌肉中地克珠利、妥曲珠利及其代谢物的残留量
林仙军1,陆春波1,包爱情1,周芷锦1,罗成江1,李孝军2,应永飞1∗
(1.浙江省兽药饲料监察所,杭州311000;2.舟山出入境检验检疫局,浙江舟山316000)
建立了一种高效液相色谱法同时测定鸡肌肉中地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜残留量的方法。试样经乙腈涡旋提取,正己烷脱脂,经碱性氧化铝小柱净化,浓缩,流动相定容,高效液相色谱分离,紫外检测器检测,以保留时间定性,以峰面积定量。结果表明:该方法在0.05~5 μg/mL浓度范围内线性关系良好,相关系数r均大于0.999。在50~500 μg/kg添加浓度范围内,方法回收率在70%~110%之间,批内和批间变异系数均小于15%。方法简便、灵敏度高、稳定性好。
地克珠利;妥曲珠利;代谢物;鸡肌肉;残留量;高效液相色谱法
地克珠利和妥曲珠利是均三嗪类抗球虫药物,具有高效、低毒、广谱的驱球虫特点,广泛应用于畜禽球虫病的防治[1]。妥曲珠利在动物体内的主要代谢产物为妥曲珠利砜和妥曲珠利亚砜。由于这类药物作用时间短,临床上通常是长期重复用药,不合理用药或未严格遵守休药期等问题,容易在动物体内产生残留,通过食物链传递,进而危害消费者健康。农业部公告235号规定了地克珠利在禽肌肉中的最高残留量为500 μg/kg,妥曲珠利在鸡肌肉中的最高残留量为100 μg/kg[2]。已有研究多以HPLC法[3-5]、毛细管电泳法[6]和LC-MS/MS法[7-9]测定动物源食品中地克珠利、妥曲珠利及代谢物的残留,由于这几种药物在紫外吸收光谱上的差异,同时分析难度较大,尚未见采用液相色谱法同时检测鸡肌肉中地克珠利、妥曲珠利及其代谢物的相关报道。本研究建立了同时检测鸡肌肉中地克珠利、妥曲珠利及其代谢物残留量的方法。
1 材料与方法
1.1 仪器与试剂 Agilent1260高效液相色谱仪(配紫外检测器)(美国安捷伦公司);3k30型冷冻离心机(Sigma公司);B-490旋转蒸发仪(BuCHI公司);MS2 minishaker旋涡混匀器(IKA公司);AG-285电子天平(Mettler公司);20通道固相萃取装置(Waters公司);TU-1810紫外可见分光光度计(北京普析通用仪器有限公司);785DMP滴定仪(瑞士万通公司);乙腈(色谱纯)、甲醇(色谱纯),默克公司;乙酸乙酯、正丙醇、正己烷、四氢呋喃为分析纯;水为超纯水;地克珠利、妥曲珠利对照品为Dr.Ehrenstorfer公司产品,纯度≥98.0%;妥曲珠利亚砜、妥曲珠利砜为WiTEGA公司产品,纯度≥98.0%;Oasis®MAX 3CC/60MG,Waters公司;Sep-Pak®Vac 12cc(2g)(碱性氧化铝固相萃取小柱),Waters公司。
1.2 对照溶液的配制 地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜标准储备液的配制(100 μg/mL):称取地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜对照品各约10 mg,精密称定,于100 mL容量瓶中,加25 mL四氢呋喃溶解后,用乙腈稀释至刻度,浓度为100 μg/mL。
地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜标准工作液(5 μg/mL):准确量取标准储备液5 mL,于100 mL量瓶中,用乙腈稀释至刻度,浓度为5 μg/mL,使用时根据需要吸取适量标准工作液用流动相稀释成标准上机液。
1.3 色谱条件 流动相A为0.3%醋酸溶液,B为甲醇,梯度洗脱程序见表1所示。色谱柱:Agilent XDB C18,粒径5 μm,4.6 mm×250 mm;检测波长250 nm;进样量20 μL;柱温25℃;流速1.0 mL/min。

表1 流动相梯度洗脱表
1.4 样品的提取与净化 称取(5±0.05)g试料,于50 mL离心管中,加乙腈10 mL,旋涡混匀,中速震荡10 min,3000 r/min离心5 min,将上清液转移到100 mL分液漏斗中;下层残渣再加乙腈10 mL重复提取一次,合并两次提取液,加20 mL正己烷萃取,静置分层,取下层液体,过预先用10 mL 85%乙腈溶液活化的碱性氧化铝固相萃取小柱,自然流出,收集流出液于100 mL鸡心瓶中,加正丙醇5 mL,50℃旋转蒸发浓缩至干,冷却,残留物用1.0 mL流动相溶解,过0.45 μm滤膜,上机测定。
2 结果
2.1 标准曲线及线性范围 准确吸取地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜标准工作液,用流动相稀释成浓度分别为0.05、0.1、0.25、0.5、1.0、2.5、5.0 μg/mL的系列混合标准溶液,将此系列混合标准溶液,依次从低浓度到高浓度进行分析,按峰面积与对应的对照溶液的质量浓度做标准曲线,见表2所示。

表2 地克珠利、妥曲珠利、妥曲珠利砜和妥曲珠利亚砜标准曲线表
2.2 典型色谱图 通过样品提取与净化方法(1.4项)和色谱条件(1.3项),得到标准溶液、鸡肌肉空白样品和鸡肌肉添加样品色谱图,分别如图1、图2、图3所示,出峰的先后顺序为妥曲珠利亚砜、妥曲珠利砜、地克珠利和妥曲珠利。

图1 标准溶液色谱图(0.5 μg/mL)
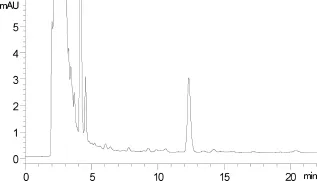
图2 鸡肌肉空白样品色谱图

图3 鸡肌肉添加样品色谱图(100 μg/kg)
2.3 方法准确度和精密度 采用标准溶液添加法,选取的3个添加浓度分别为50、100、500 μg/kg,符合残留检测的要求,其中50 μg/kg的浓度点大于10倍信噪比,作为定量限。每个浓度5个样品,重复3批次,求回收率、批内变异系数、批间变异系数。测定结果见表3。
3 讨论
3.1 检测波长的选择 为了确定最佳检测波长,采用紫外分光光度法进行紫外扫描。分别用乙腈稀释地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜对照品溶液至约5 μg/mL,在200~400 nm的波长范围进行扫描,结果表明,地克珠利在240 nm和280 nm的波长处有最大吸收,妥曲珠利在240 nm的波长处有最大吸收,而妥曲珠利砜和妥曲珠利亚砜在250 nm的波长处有最大吸收。综合考虑,选择250 nm作为检测波长,地克珠利、妥曲珠利、妥曲珠利砜和妥曲珠利亚砜响应值均较多,溶剂峰对色谱峰的干扰较小。
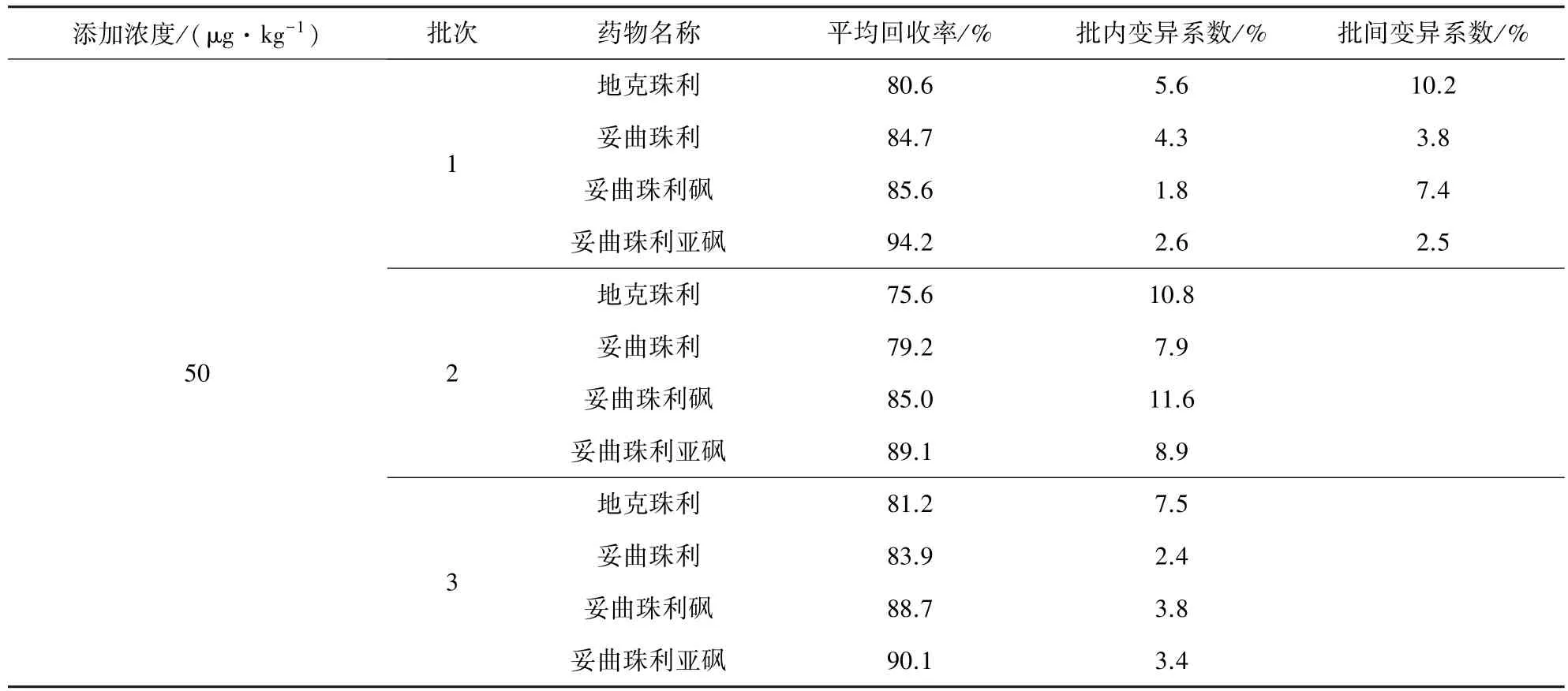
表3 鸡肌肉中地克珠利、妥曲珠利、妥曲珠利砜及妥曲珠利亚砜的添加回收率测定结果表(n=5)

续表
3.2 流动相的选择 考察了乙腈-水、乙腈-磷酸溶液、乙腈-醋酸溶液、甲醇-水、甲醇-磷酸溶液和甲醇-醋酸溶液等体系作为流动相进行分析,从实验结果得知,乙腈-水、甲醇-水作为流动相峰形较差,其余溶液作为流动相的条件下色谱峰峰形和分离度均较好,但乙腈体系基线不易平稳,对低浓度检测会产生干扰;甲醇-醋酸溶液体系色谱基线稳定,没有干扰,满足检测需要。在实际样品检测中发现,试样溶液在4种药物峰全部出来以后还有2个较大的杂质峰,可能会影响到下一针样品。因此,增加梯度洗脱程序,对杂质峰进行洗脱,确保样品不受之前进样的样品干扰。
3.3 提取溶剂的选择 研究了乙酸乙酯、乙腈等提取溶剂,乙酸乙酯和乙腈提取效率均较高。乙酸乙酯提取液较易旋转蒸发至干,但脂溶性太强,复溶的溶液粘度大,浑浊,难以过柱,影响后续净化,净化后亦不能去除干扰;乙腈作为提取溶剂,能有效沉淀蛋白,方便正己烷脱脂,为后续固相萃取净化提供方便。
3.4 固相萃取柱条件的选择 根据查阅的资料,选择了MAX、碱性氧化铝等小柱作为净化柱进行试验。MAX固相萃取小柱可以净化样品,但是过柱程序复杂,增加旋转蒸发的次数,大大增加检测时间和不确定性因素;而碱性氧化铝小柱,过柱方式简便,回收率和净化效果均较好。考虑到乙腈提取液中水分的含量对回收率有很大影响,因此,对乙腈提取液中的水分进行了测定,比较了回收率和水分之间的关系。用乙腈10 mL提取5 g试料的提取液水分的含量约20%,重复提取一次,合并提取液,水分约15%。根据过柱回收率结果研究,水分在10%~30%范围,过碱性氧化铝,回收率均在90%左右,净化效果对应水分高低略有差异,提取液中水分低于10%,回收率会降低,水分高于30%,则杂质太多,干扰检测。用乙腈10 mL提取2次,水分含量比较合适。试验了不同批次的鸡肌肉样品,提取液水分均在这个范围之内。
4 结论
本文建立了鸡肌肉中地克珠利、妥曲珠利及其代谢物残留量检测的方法。方法采用乙腈提取,提取液脱脂后直接过固相萃取小柱进行净化,过柱的要求低,操作简便,净化效果和回收率较好并稳定,能满足检验需要。该方法明显节约检验时间和试剂用量,有利于提高检测效率。
[1] 冯秀娟,刘英龙,王加才,等.广谱抗球虫药-妥曲珠利[J].中国禽业导刊,2006,23(1):29.
[2] 中华人能共和国农业部公告235号.动物性食品中兽药最高残留限量[S].
[3] 施祖灏,朱良强,卢运站,等.鸡组织中地克珠利和妥曲珠利HPLC检测方法的建立[J].中国兽医学报,2009,29(1):79-81,109.
[4] 郭永刚,江善祥.妥曲珠利在肉鸡组织中残留的研究[J].畜牧兽医,2007,39(7):37-39.
[5] 杨丽萍,高淑霞,张秀玲,等.HPLC法检测家兔血浆中地克珠利含量[J].中国兽药杂志,2011,45(10):25-27.
[6] 施祖灏,陆俊贤,葛庆联,等.高效毛细管电泳法同时检测地克珠利和妥曲珠利的含量[J].中国兽药杂志,2008,42(9).13-16.
[7] 宫小明,孙军,董静,等.高效液相色谱-串联质谱法测定猪肉中阿维菌素类、地克珠利、妥曲珠利及其代谢物残留[J].色谱,2011,29(3):217-222.
[8] Mortier L,Daeseleire E,Peteghem C V.Determination of the coccidiostat diclazuril in poultry feed and meat by liquid chroma⁃tography-tandem mass spectrometry[J].Analytica Chimica Acta,2005,529:229-234.
[9] Olejnik M,Szprengier-Juszkiewicz T,Jedziniak P.Multi-residue confirmatory method for the determination of twelve coc cidiostats inchickenliverusingliquidchromatographytandemmass spectrometry[J].Journal of Chromatography A,2009,1216(46):8141-8148.
(编辑:侯向辉)
Determination of Diclazuril,Toltrazuriland and Its Metabolites in Chicken Muscle by HPLC
LIN Xian-jun1,LU Chun-bo1,BAO Ai-qing1,ZHOU Zhi-jin1,LUO Cheng-jiang1,LI Xiao-jun2,YING Yong-fei1∗
(1.Zhejiang Province Supervisory Institute of Veterinary Drug and Feed,Hangzhou 311000,China;2.Zhoushan Entry-exit Inspection and Quarantine Bureau,Zhoushan,Zhejiang 316000,China)
A method was established for simultaneously determination of diclazuril,toltrazuril,toltrazuril sulphone and toltrazuil sulphoxide in chicken muscle by HPLC.The analytes were extracted with acetonitrile,defatted with n-hexane,and then clean up by alumina solid-phase extraction cartridges.The eluent was evaporated by nitrogen flow to dryness and dissolved the residues with mobile phase.HPLC was employed for separation and UV detector was used for detecting directly.Qualitative analysis was done by retention time and quantitative analysis by peak area respectively.The results showed that good linear relationship were obtained in the range of 0.05~5 μg/mL,with the correlation coefficients were all above 0.999.The average recoveries ranged from 70%to 110%of the spiked level at 50~500 μg/kg,and the intra-assay coefficient and the inter-assay coefficient of variation were all below 15%.The method was simple,sensitive and stable.
diclazuril;toltrazuril;metabolite;chicken muscle;residues;HPLC
2015-01-19
A
1002-1280(2015)03-0053-05
S859.84
农业部行业标准制定与修订项目(农财发201461(96))
林仙军,畜牧师,从事兽药和畜产品质量安全检验工作。
应永飞。E-mail:yyf1001@163.com
